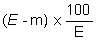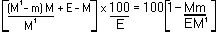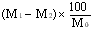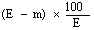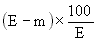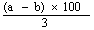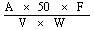S.I. No. 249/1978 -- Fertilisers, Feeding Stuffs and Mineral Mixtures (Methods of Analysis) Regulations, 1978.
|
S.I. No. 249/1978: FERTILISERS, FEEDING STUFFS AND MINERAL MIXTURES (METHODS OF ANALYSIS) REGULATIONS, 1978. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
FERTILISERS, FEEDING STUFFS AND MINERAL MIXTURES (METHODS OF ANALYSIS) REGULATIONS, 1978. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
I, JAMES GIBBONS, Minister for Agriculture in exercise of the powers conferred on me by Section 11 of the Fertilisers, Feeding Stuffs and Mineral Mixtures Act, 1955 (No. 8 of 1955) (as adapted by the Agriculture and Fisheries (Alteration of Name of Department and Title of Minister) Order, 1977 ( S.I. No. 31 of 1977 )) hereby make the following regulations: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. These Regulations may be cited as the Fertilisers, Feeding Stuffs and Mineral Mixtures (Methods of Analysis) Regulations, 1978. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. These Regulations shall come into operation on the 1st day of October, 1978. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. In these Regulations "the Principal Regulations" means the Fertilisers, Feeding Stuffs and Mineral Mixtures Regulations, 1957 ( S.I. No. 264 of 1957 ). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. The Principal Regulations are hereby amended by the substitution, for Article 10, of the Articles set out in Part I of the Schedule to these Regulations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. The Principal Regulations are hereby further amended-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(a) by the substitution, for Form F.F.2 in the Third Schedule, of the form set out in Part II of the Schedule to these Regulations, and |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(b) by the substitution, for Form F.F.3 in the Third Schedule, of the form set out in Part III of the Schedule to these Regulations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
SCHEDULE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
PART I. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The manner in which a sample of a feeding stuff, compound feeding stuff or mineral mixture shall be analysed for the purposes of the Act shall be as specified in the subsequent paragraphs of this Part of the Schedule. The main divisions of this Part of the Schedule are as follows: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. General Provisions. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Determination of moisture. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Determination of moisture in animal and vegetable fats and oils. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Determination of moisture in milk products and milk replacers. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Determination of moisture in mineral substances and mixtures composed predominantly of mineral substances. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Determination of crude oils and fats. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination of crude protein. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
8. Determination of true protein. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
9. Determination of crude fibre. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
10. Determination of betacarotene. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
11. Determination of total phosphorus (a) volumetric method. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
12. Determination of total phosphorus (b) photometric method. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
13. Determination of crude ash. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
14. Determination of ash which is insoluble in HC1. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
15. Determination of calcium. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
16. Determination of iodine. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
17. Determination of water-soluble chlorides. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
18. Determination of cobalt. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
19. Determination of copper. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
20. Determination of iron. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
21. Determination of manganese. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
22. Determination of magnesium. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. GENERAL PROVISIONS. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.1 Preparation of the sample for analysis. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.1.1. The sample shall be divided into two parts. One part shall be taken as it is for macroscopic and microscopic determinations and also the determination of moisture. The other part shall be prepared as follows for chemical analysis. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.1.2 Mix this part carefully on a clean dry surface. (If this part is very moist it must be pre-dried to bring the moisture content down to a level between 8--12%. To do this, dry the sample at a suitable temperature for an adequate length of time). Divide it by means of a divider or by hand using the quartering method (which consists of taking portions in turn from two opposite sections). Reduce it by either of these methods to approximately 100 g and, if necessary, crush the reduced portion so that it will pass through a 1 mm round mesh sieve. Transfer this portion immediately to a dry container with an airtight fitting. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.2 Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
All reagents used should be of analytical quality. References to water mean distilled or de-ionised water. Where a solution of a reagent is mentioned without any other indication this means a solution in distilled or de-ionised water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.3 Expression of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The result given in the certificate of analysis shall be the average value obtained on the basis of at least two tests. Subject to special provisions, it shall be expressed as a proportion by weight of the original sample as it was when it reached the State Laboratory. The result must not be given to more significant figures than the accuracy of the method of analysis allows. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. DETERMINATION OF MOISTURE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the moisture content of feeding stuffs, other than animal and vegetable fats and oils, milk products and milk replacers, mineral substances and mixtures composed predominantly of mineral substances, or oilseeds and oleaginous fruit as defined in Council Regulation No. 136/66/EEC of 22 September 1966(1). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 OJ No. 127, 30. 9. 1966, P. 3025/66. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is desiccated under specified conditions which vary according to the nature of the feeding stuffs. The loss in mass is determined by weighing. It is necessary to carry out preliminary drying when dealing with solid feeding stuffs which have a high moisture content. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Crusher of non moisture-absorbing material which is easy to clean, allows rapid, even crushing without producing any appreciable heating, prevents contact with the outside air as far as possible and meets the requirements laid down in 4.1.1 and 4.1.2 (e.g. hammer or water-cooled micro-crushers, collapsible cone mills, slow motion or cog- wheeled crushers). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Analytical balance, accurate to 0·5 mg. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Dry containers of non-corrodible metal or of glass with lids ensuring airtight closure; working surface allowing the test sample to be spread at about 0·3 g/cm2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Electrically heated isothermal oven (± 1°C) properly ventilated and ensuring rapid temperature regulation.2 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2For the drying of cereals, flour, groats and meal, the oven must have a thermal capacity such that, when pre-set at 131°C it will return to that temperature in less than 45 minutes after the maximum number of test samples have been placed inside to dry simultaneously. Ventilation must be such that, when as many samples of common wheat as it can contain are dried for 2 hours, the results differ from those obtained after 4 hours of drying by less than 0·15%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Adjustable electrically heated vacuum oven fitted with an oil pump and either a mechanism for introducing hot, dried air or a drying agent (e.g. calcium oxide). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Dessicator with a thick perforated metal or porcelain plate, containing an efficient drying agent. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
N.B. The operations described in this section must be carried out immediately after opening the packages of samples. Analysis must be carried out at least in duplicate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Preparation. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1.1 Feeding stuffs other than those coming under 4.1.2 and 4.1.3 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Take at least 50 g of the sample. If necessary, crush or divide in such a way as to avoid any variation in moisture content (see 6). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1.2 Cereals and groats |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Take at least 50 g of the sample. Grind into particles of which at least 50 per cent will pass through a 0·5 mm mesh sieve and will leave no more than 10 per cent reject on a 1 mm round-meshed sieve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1.3 Feeding stuffs in liquid or paste form, feeding stuffs predominantly composed of oil |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 10 mg, approximately 25 g of the sample. Add an appropriate quantity of anhydrous sand weighed to the nearest 10 mg and mix until a homogeneous product is obtained. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Drying. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2.1 Feeding stuffs other than those coming under 4.2.2 and 4.2.3 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 0·5 mg a container (3.3) with its lid. Weigh into the weighed container, to the nearest mg, about 5 g of the sample and spread evenly. Place the container, without its lid, in the oven preheated to 103°C. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible. Leave to dry for 4 hours reckoned from the time when the oven temperature returns to 103°C. Replace the lid on the container, remove the latter from the oven, leave to cool for 30--45 minutes in the desiccator (3.6) and weigh to the nearest mg. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
For feeding stuffs composed predominantly of oil, dry in the oven for an additional 30 minutes at 130°C. Cool in a desiccator and weigh. The difference between the two weighings must not exceed 0·1 per cent of moisture. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2.2 Cereals, flour, groats and meal |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 0·5 mg, a container (3.3) with its lid. Weigh into the weighed container, to the nearest mg, about 5 g of the crushed sample and spread evenly. Place the container, without its lid in the oven (3.4) preheated to 130°C. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible. Leave to dry for 2 hours reckoned from the time when the oven temperature returns to 130°C. Replace the lid on the container, remove the latter from the oven, leave to cool for 30--45 minutes in the desiccator (3.6) and weigh to the nearest mg. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2.3 Compound feeding stuffs containing more than 4 per cent of sucrose or lactose; straight feeding stuffs such as locust beans, hydrolized cereal products, maltculms, dried beet pulp, fish and sugar solubles; compound feeding stuffs containing more than 25 per cent of mineral salts including water of crystallisation |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 0·5 mg, a container (3.3) with its lid. Weigh into the weighed container, to the nearest mg, about 5 g of the sample and spread evenly. Place the container, without its lid, in the vacuum oven (3.5) preheated to between 80--85°C. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Bring the pressure up to 100 Torr and leave to dry for 4 hours at this pressure, either in a current of hot, dry air or using a drying agent (about 300 g for 20 samples). In the latter instance, disconnect the vacuum pump when the prescribed pressure has been reached. Reckon drying time from the moment when the oven temperature returns to 80--85°C. Carefully bring the oven back to atmospheric pressure. Open the oven, place the lid on the container immediately, remove the container from the oven, leave to cool for 30--45 minutes in the desiccator (3.6) and weigh to the nearest mg. Dry for an additional 30 minutes in the vacuum oven at 80--85°C and reweigh. The difference between the two weighings must not exceed 0·1 per cent of moisture. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Preliminary drying. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3.1 Feeding stuffs other than those coming under 4.3.2 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Solid feeding stuffs with a high moisture content which makes crushing difficult must be subjected to preliminary drying as follows: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 10 mg, approximately 50 g of uncrushed sample (compressed or agglomerated feeding stuffs may be roughly divided if necessary) in a suitable container (eg. a 20 x 12 cm aluminium plate with a 0·5 cm rim). Leave to dry in an oven from 60--70°C until the moisture content has been reduced to between 8--12 per cent. Remove from the oven, leave to cool uncovered in the laboratory for 1 hour and weigh to the nearest 10 mg. Crush immediately as indicated in 4.1.1 and dry as indicated in 4.2.1 or 4.2.3 according to the nature of the feeding stuff. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3.2 Cereals |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Grain with a moisture content of over 17 per cent must be subjected to preliminary drying as follows: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 10 mg, 50 g of unground grain in a suitable container (e.g. a 20 x 12 cm aluminium plate with a 0·5 cm rim). Leave to dry for 5--7 minutes in an oven at 130°C. Remove from the oven, leave to cool uncovered in the laboratory for 2 hours and weigh to the nearest 10 mg. Grind immediately as indicated in 4.1.2 and dry as indicated in 4.2.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The moisture content, as a percentage of the sample, is calculated by using the following formulae: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Drying without preliminary drying. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Drying with preliminary drying. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample should not exceed 0·2% in absolute value. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Observation. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
If crushing proves necessary and if this is seen to alter the moisture content of the product, the results of the analysis of the components of the feeding stuff must be corrected on the basis of the moisture content of the sample in its initial state. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. DETERMINATION OF MOISTURE IN ANIMAL AND VEGETABLE FATS AND OILS |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the moisture and volatile substances content of animal and vegetable fats and oils. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is dried to constant weight at 103°C. The loss in mass is determined by weighing. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Flat-bottomed dish, of a corrosion-resistant material, 8--9 cm in diameter and approximately 3 cm high. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Mercury thermometer with a strengthened bulb and expansion tube at the top end, graduated from approximately 80°C to at least 110°C, and approximately 10 cm in length. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Sand bath or electric hot-plate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Desiccator, containing an efficient drying agent. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Analytical balance. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 20 g of the homogenized sample into the dry, weighed dish (3.1) containing the thermometer (3.2). Heat on the sand bath or hot-plate (3.3), stirring continuously with the thermometer, so that the temperature reaches 90°C in about 7 minutes. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Reduce the heat, watching the frequency with which bubbles rise from the bottom of the dish. The temperature must not exceed 105°C. Continue to stir, scraping the bottom of the dish, until bubbles stop forming. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
In order to ensure complete elimination of moisture, reheat several times to 103°C ± 2°C, cooling to 93°C between successive heatings. Then leave to cool to room temperature in the desiccator (3.4) and weigh. Repeat this operation until the loss in mass between two successive weighings no longer exceeds 2 mg. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
N.B. An increase in the mass of the sample after repeated heating indicates an oxidation of the fat, in which case calculate the result from the weighing carried out immediately before the mass began to increase. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The moisture content, as a percentage of the sample, is given by the following formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
where: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Results lower than 0·05% must be recorded as "lower than 0·05%". |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference in moisture between the results of two parallel determinations carried out on the same sample must not exceed 0·05%, in absolute value. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. DETERMINATION OF MOISTURE IN MILK PRODUCTS AND MILK REPLACERS. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the moisture content of milk products and milk replacers. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is dried for 3 hours at 102°C±2°C. The loss in mass is determined by weighing. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Analytical balance. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Flat bottomed dishes of non-corrodible metal or of glass with lids ensuring airtight closure; working surface allowing the test sample to be spread at about 0·3 g/cm2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Electrically heated isothermal oven (± 1°C) properly ventilated and ensuring rapid temperature regulation. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Desiccator containing an efficient drying agent. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place dishes (3.2) in drying oven at 102°C (3.3) for approximately 30 minutes. Cool in a desiccator (3.4) and weigh accurately. Weigh, to the nearest mg, approximately 3 g of sample into the dish. Transfer to oven (102°C) and leave for 3 hours. Remove to a desiccator (3.4). Cool and weigh. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The moisture content, as a percentage of the sample, is calculated by using the following formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. DETERMINATION OF MOISTURE IN MINERAL SUBSTANCES AND MIXTURES COMPOSED PREDOMINANTLY OF MINERAL SUBSTANCES. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the moisture content of mineral substances and mixtures composed predominantly of mineral substances. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is dried to a constant weight at 100°C and the loss in mass is determined by weighing. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Analytical balance. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Electrically heated isothermal oven (± 1°C) properly ventilated and ensuring rapid temperature regulation. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Desiccator, containing an efficient drying agent. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Dry containers of non-corrodible metal or glass with air tight lids. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh container (3.4) with its lid to the nearest 0·5 mg. Weigh into the container, to the nearest mg, about 10 g of sample and spread evenly. Place container without its lid in an oven (3.2) preheated to 100°C. Dry until two successive weighings, after cooling in a desiccator (3.3) at intervals of not less than 3 hours in the oven, show an increment of loss of not more than 0·2 per cent of the original weight. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The moisture content, as a percentage of the sample, is calculated by using the following formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. DETERMINATION OF CRUDE OILS AND FATS. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and Scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of crude oils and fats in feeding stuffs. It does not cover the analysis of the oil seeds and oleaginous fruit defined in Council Regulation 136/66/EEC of 22 September, 1966. The determination of the oil content of those products is described in Annex V to Commission Regulation (EEC) No. 1470/68 of 23 September 1968.1 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 O.J. No. L239 28/9/1968 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Either one of two methods may be used, depending on the nature of the feeding stuff. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.1 Method A (extraction by light petroleum): applicable to all feeding stuffs other than those coming under 1.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.2 Method B: applicable to feeding stuffs from which oils and fats cannot be totally extracted with light petroleum without prior hydrolysis, to feeding stuffs of animal origin, glutens, dried potato pulps, dried brewing and distilling dreys and waste, dried yeasts, waste from biscuits, bread and cooked foods, milk products and feeding stuffs containing a high proportion of such products (at least 40%) and to compound feeding stuffs enriched with fats. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2.1 Method A: The oils and fats are extracted with light petroleum. The solvent is distilled off and the residue dried and weighed. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2.2 Method B: The sample is hydrolised when hot with hydrochloric acid. The solution is cooled and filtered. The residue is washed and dried and extracted with light petroleum using Method A. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Light petroleum, boiling range 40--60°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Anhydrous sodium sulphate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Hydrochloric acid, 3 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Filtration aid, e.g. Kieselgur, Hyflo-supercel. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Carbon tetrachloride. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Soxhlet-type extractor or equivalent apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Explosion-proof heating apparatus with temperature control. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Vacuum drying oven (less than 100 Torr). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Method A: (see 7.1) |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample and mix with 2--3 g (or more, if necessary) of anhydrous sodium sulphate (3.2). Place the mixture in an extraction thimble free from oils and fats and cover with a fat-free wad of cottonwool. (The mixing may be carried out in the thimble). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place the thimble in an extractor (4.1) and extract for 6 hours with light petroleum (3.1). If a Soxhlet-type extractor is used, regulate the heating to obtain at least 15 siphonings per hour. Collect the extract in a dry, weighed flask containing fragments of pumice stone1. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Distil off the solvent and dry the evaporation residue for 1½ hours in the vacuum drying oven (4.3) at 75°C. Cool in a desiccator and weigh. Dry again for 30 minutes to ensure that the weight of the oil and fat remains constant (loss in weight must be less than 1 mg). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Method B |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 2·5 g of the sample (see 7.2) and place in a 400 ml beaker or a 300 ml Erlenmeyer flask. Add 100 ml of hydrochloric acid 3 N (3.3) and fragments of pumice stone. Cover the beaker with a watch glass or fit the Erlenmeyer flask with a reflux condenser. Bring the mixture to a gentle boil over a low flame or a hot plate and keep it there for 1 hour. Do not allow the product to stick to the sides of the container. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Cool and add a quantity of filtration aid (3.4) sufficient to prevent any loss of oil and fat during filtration. Filter through a moistened, fat-free, double filter paper. Wash the residue in cold water until the acid reaction has ceased. Check that the filtrate does not contain any oil or fats. Their presence in the filtrate indicates that the sample must be extracted with light petroleum, using the method given in 5.1, before hydrolysis. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place the double filter paper containing the residue on a watch glass and dry for 1½ hours in the oven at 95°C--98°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place the double filter paper and the dry residue in an extraction thimble, extract with light petroleum and proceed as indicated in the second paragraph of 5.1. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability. The difference between the results of two parallel determinations carried out on the same sample should not exceed 0·3% of oil or fat. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 For products with a high content of oils and fats and which are difficult to crush or unsuitable for drawing a homogeneous reduced test sample, proceed as follows. Weigh 20 g of the sample to the nearest mg and mix with 10 g or more of anhydrous sodium sulphate (3.2). Extract with light petroleum (3.1) as indicated in 5.1. Make up the extract obtained to 500 ml with carbon tetrachloride (3.5) and mix. Take 50 ml of the solution and place in a small, dry, weighed flask containing fragments of pumice stone1. Distil off the solvent, dry and proceed as indicated in the last paragraph of 5.1. Eliminate the solvent from the extraction residue left in the thimble and crush the residue to a fineness of 1 mm. Return the product to the extraction thimble (do not add sodium sulphate), extract with light petroleum and proceed as indicated in the second and third paragraphs of 5.1. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 Where the oil or fat has to undergo subsequent quality tests, replace the fragments of pumice stone by glass beads. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the results as a percentage of the sample, taking into account the aliquot part used for the first extraction, by using the following formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(10a + b) x 5 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 For products low in oils and fats the test sample may be increased to 5 g. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. DETERMINATION OF CRUDE PROTEIN. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the crude protein content of feeding stuffs on the basis of the nitrogen content, determined according to the Kjeldahl method. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is digested by mineral acid. The acid solution is alkalized with a sodium hydroxide solution. The ammonia released is removed by distillation and collected in a measured quantity of sulphuric acid, the excess of which is titrated with a solution of sodium hydroxide. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Potassium sulphate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Catalyst: cupric oxide CuO or crystallized cupric sulphate CuSO4. 5H2O or mercury or mercuric oxide HgO. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Granulated zinc. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Sulphuric acid, d: 1.84 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sulphuric acid, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Sulphuric acid, 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Methyl red indicator: dissolve 300 mg of methyl red in 100 ml of 95--96% (v/v) ethanol. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Sodium hydroxide solution, 40% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Sodium hydroxide solution, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Sodium hydroxide solution, 0.25 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Sodium sulphide, saturated solution. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Sodium thiosulphate solution, 8% (w/v) Na2S2O3. 5H2O. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Granulated pumice stone, washed in hydrochloric acid and ashed. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 Where the oil or fat has to undergo subsequent quality tests, replace the fragments of pumice stone by glass beads. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Apparatus for digestion by combustion and for distillation by the Kjeldahl method (see item 7.1). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Digestion |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 1 g of sample and place in the flask of the digestion apparatus. Add 10 g of potassium sulphate (3.1), an appropriate quantity of catalyst (3.2) (0.3--0.4 g of cupric oxide or 0.9--1.2 g of cupric sulphate or a drop of mercury or 0.6--0.7 g of mercuric oxide), 25 ml of sulphuric acid (3.4) and a few granules of pumice stone (3.13). Mix. Heat the flask moderately at first, shaking from time to time, until the mass has carbonized and the foam has disappeared; then heat more intensively until the liquid is boiling steadily. Prevent the sides from becoming overheated and organic particles from sticking to them. When the solution becomes clear and colourless (or light green if a copper-based catalyst is used), continue to boil for another hour, then leave to cool. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Distillation |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Carefully add 250--350 ml of water, stirring all the while to dissolve the sulphates completely; leave to cool. Add a few granules of zinc (3.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place in the collecting flask of the distillation apparatus an exactly measured quantity of 25 ml of sulphuric acid 0.1 N (3.5) or 0.5 N (3.6) depending on the presumed nitrogen content (see item 7.2), and add a few drops of methyl red indicator (3.7). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Connect the distillation flask to the condenser of the distillation apparatus and immerse the end of the condenser in the liquid contained in the collecting flask to a depth of at least 1 cm (see item 7.3). Slowly pour 100 ml of 40% sodium hydroxide solution (3.8) into the distillation flask through the dropping funnel. If a mercury-based catalyst has been used, also add either 10 ml of sodium sulphide solution (3.11), or 25 ml of sodium thiosulphate solution (3.12). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Heat the flask in such a way that approximately 150 ml of liquid is distilled in 30 minutes. At the end of this time, check the pH of the resulting distillate with litmus paper. If the reaction is alkaline, continue distillation. Discontinue when the distillate becomes neutral to litmus paper. During distillation keep the colouration under observation and shake the contents of the collecting flask from time to time. If the liquid turns yellow, immediately add an exactly measured volume of sulphuric acid 0.1 N (3.5) or 0.5 N (3.6). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Titration |
|||||||||||||||||||||||||||||||||||||||||||||||
|
In the collecting flask titrate the excess sulphuric acid with sodium hydroxide solution 0.1 N (3.9) or 0.25 N (3.10), depending on the normality of the sulphuric acid used, until the colour turns pale yellow. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Verification of the method |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To establish whether the reagents are free from nitrogen, carry out a blank test (distillation and titration) omitting the sample to be analysed. To check that the apparatus is working properly and that the correct application of the method is used carry out the analysis (digestion, distillation and titration) on 1.5--2.0 g of acetanilide (m.p. 114°C: %N: 10.36) in the presence of 1 g of nitrogen-free sucrose; 1 g of acetanilide corresponds to 14.80 ml of sulphuric acid 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Determine the volume of sulphuric acid neutralised. 1 ml of sulphuric acid 0.1 N corresponds to 1.4 mg of nitrogen. Multiply the quantity of nitrogen by the factor 6.25. Express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
--0·2%, in absolute value, for crude protein contents of less than 20%; |
|||||||||||||||||||||||||||||||||||||||||||||||
|
--1·0%, relative to the higher result, for contents of not less than 20% and not more than 40%; |
|||||||||||||||||||||||||||||||||||||||||||||||
|
--0·4%, in absolute value, for contents of more than 40%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Certain apparatus requiring transference between digestion and distillation may be used. If such apparatus is used, the transfer must be carried out without loss. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 For products with a low nitrogen content, the volume of sulphuric acid 0.1 N to be placed in the collecting flask may be reduced, if necessary, to 10 or 15 ml and made up to 25 ml with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.3 If the flask of the distillation apparatus is not fitted with a dropping funnel, add the sodium hydroxide immediately before connecting the flask to the condenser, pouring the liquid slowly down the sides of the condenser so that it does not mix with the acid solution. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
8. DETERMINATION OF TRUE PROTEIN. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the true protein content of feeding stuffs. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The protein is precipitated by cupric hydroxide. The copper protein precipitate is digested by mineral acid. The acid solution is made alkaline by a sodium hydroxide solution. The ammonia released is removed by distillation and collected in a measured quantity of sulphuric acid, the excess of which is titrated by a solution of sodium hydroxide. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Potassium sulphate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Catalyst: cupric oxide CuO, or crystallised cupric sulphate CuSO4 . 5H2O, or mercuric oxide HgO. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Zinc granulated. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Sulphuric acid, d: 1.84. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sulphuric acid, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Sulphuric acid, 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Methyl red indicator: dissolve 300 mg methyl red in 100 ml ethanol (95--96% v/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Sodium hydroxide solution, 40% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Sodium hydroxide solution, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Sodium hydroxide solution, 0.25 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Sodium sulphide solution, saturated. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Sodium thiosulphate solution, 8% (w/v) Na2 S2 O3. 5H2O. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Granulated pumice stone, washed in hydrochloric acid and calcined. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Cupric hydroxide reagent: dissolve 100 g of crystalline copper sulphate in 5 litres of water. Add 2.5 ml of glycerol and then 10% sodium hydroxide solution until the liquid is slightly alkaline. Filter the mixture and mix the precipitate in a mortar containing 5 ml of glycerol per litre. Wash the precipitate by decantation or filtration until the washings are no longer alkaline. Again mix the precipitate in a mortar with water containing 10% of glycerol until a uniform gelatinous mass measurable by pipette is produced. The quantity of cupric hydroxide in 5 ml is determined by diluting to 50 ml with water, filtering, washing, igniting and weighing as cupric oxide. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Apparatus for mineral acid digestion and distillation according to Kjeldahl's method (see item 7.1). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Precipitation of protein. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 0.7 g of sample and mix with 100 ml of water in a beaker. If the sample is rich in starch heat on a steam bath for 10 minutes, otherwise heat the mixture to boiling. Add a quantity of reagent (3.14) containing about 0.5 g cupric hydroxide. Allow to cool and transfer the precipitate quantitatively to a nitrogen-free filter paper. Thoroughly wash the precipitate and filter paper with cold water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Wet combustion |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Transfer precipitate and filter paper to a Kjeldahl flask, add 10 g potassium sulphate (3.1), an appropriate quantity of catalyst (3.2) (0.3--0.4 g cupric oxide, or 0.9--1.2 g cupric sulphate, or a drop of mercury, or 0.6--0.7 g mercuric oxide), 25 ml sulphuric acid (3.4) and a few grains of pumice stone (3.13). Mix. Heat the flask moderately at first, shaking from time to time, until the mass is carbonised and the froth has disappeared; then increase the heat and bring the liquid to a steady boil. Avoid overheating which may cause the organic particles to stick to the side of the flask. When the solution appears clear and colorless (light green in the presence of catalysts with a copper base) continue boiling for another hour. Then allow to cool. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Distillation |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Add 250--350 ml water with care and stirring all the while to dissolve the sulphates completely; allow to cool. Then add a few grains of zinc (3.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place an accurately measured volume of 25 ml sulphuric acid 0.1 N (3.5) or 0.5 N (3.6) in the collecting flask of the distillation apparatus, according to the presumed level of nitrogen and add a few drops of methyl red indicator (3.7). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Connect the flask to the condenser of the distillation apparatus and place the end of the condenser at least 1 cm below the surface of the liquid in the collecting flask. Slowly pour 100 ml of 40 per cent sodium hydroxide solution (3.8) into the flask using the funnel with stopcock. If a mercury-based catalyst has been used, add to the flask either 10 ml sodium sulphide solution (3.11), or 25 ml sodium thiosulphate solution (3.12). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Heat the flask so that approximately 150 ml of the liquid is distilled in 30 minutes. At the end of this time, check the pH of the resulting distillate with litmus paper. If the reaction is alkaline, continue the distillation. Discontinue distillation when the distillate appears neutral to litmus paper. During the distilling process, swirl the contents of the collecting flask from time to time and watch the color. If this turns yellow, add immediately an exactly measured volume of sulphuric acid 0.1 N (3.5) or 0.5 N (3.6). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Titration |
|||||||||||||||||||||||||||||||||||||||||||||||
|
In the collecting flask titrate the excess sulphuric acid with sodium hydroxide solution 0.1 N (3.9) or 0.25 N (3.10), according to the normality of the sulphuric acid used, until the color turns pale yellow. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Blank test |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To establish that the reagents are nitrogen free, carry out a blank test (distillation and titration) without the sample for analysis. To establish that the apparatus is working properly and that the correct application of the method is used, carry out the analysis (wet combustion, distillation and titration) on 1.5--2.0 g acetanilide (melting point, 114°C; N, 10.36 per cent) in the presence of 1 g nitrogen-free sucrose: 1 g acetanilide consumes 14.80 ml 0.5 N sulphuric acid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Determine the volume of sulphuric acid neutralised. 1 ml 0.1 N sulphuric acid corresponds to 1.4 mg nitrogen. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Multiply the quantity of nitrogen by the factor 6.25. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Certain apparatus may require a decantation between wet combustion and distillation. In this case, decantation must be carried out without loss. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 If the sample is of a substance rich in alkaline phosphates, add 1 or 2 ml of 10% solution of ammonia-free sodium aluminium sulphate to the mixture before the addition of the cupric hydroxide, in order to prevent the formation of cupric phosphate and free alkali and the consequent partial solution of the copper-protein precipitation and alkaline liquid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
9. DETERMINATION OF CRUDE FIBRE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine, in feeding stuffs, the content of fat-free organic substances which are insoluble in acid and alkaline media and are conventionally described as crude fibre. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample, defatted where necessary, is treated successively with boiling solutions of sulphuric acid and potassium hydroxide of specified concentrations. The residue is separated by filtration in the presence of asbestos, washed, dried, weighed and ashed at 900°C. The loss of weight resulting from ashing corresponds to the crude fibre present in the test sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Sulphuric acid, 0.26 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Treated asbestos: add to asbestos of the type used with a Gooch crucible approximately 5 times its weight of dilute hydrochloric acid (1 volume hydrochloric acid, d: 1.19 + 3 volumes of water). Boil the mixture for approximately 45 minutes, leave to cool and filter through a Buchner funnel. Wash the residue first with water until the washing water is free from acid, and then with acetone (3.6). Dry the asbestos in the drying oven and then ash for 2 hours at 900°C. Leave to cool and keep in a stoppered flask. Asbestos treated in this way may be used several times. It must meet the specifications given in 5 regarding the blank test. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Antifoam (e.g. silicone). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Potassium hydroxide solution 0.23 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Hydrochloric acid 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Acetone. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Light petroleum, boiling range 40--60°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Beakers of at least 600 ml capacity, with measuring marks at the 200 ml level. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Porcelain discs approximately 80 mm in diameter and approximately 4 mm thick, perforated with approximately 32 holes, each approximately 4 mm in diameter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Rubber-stoppered vacuum flasks of approximately 2 litre capacity, with measuring marks at the 800 ml level and fitted with glass funnels 120 mm in diameter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Filter plates approximately 40 mm in diameter and approximately 4 mm thick, with slanting edges to fit the cone of the funnel (4.3), perforated with approximately 16 holes, each approximately 4 mm in diameter, and covered by a wire mesh, the mesh size being approximately 1 mm. Both plates and wire mesh must be resistant to acids and alkali. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Platinum or silica ashing crucibles. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Thermostatically controlled electric muffle-furnace. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Desiccator. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Asbestos filter: suspend 2.0 g asbestos (3.2) in 100 ml water. Filter under vacuum over a filter plate covered with a wire mesh (4.4) and placed in the funnel of a vacuum flask (4.3). Collect the filtrate and filter once more through the same filter. Discard the filtrate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 3 g of the sample and 2 g treated asbestos (3.2) into a beaker (4.1), add 200 ml sulphuric acid (3.1) and a few drops of antifoam (3.3). Bring rapidly to the boil and leave to boil for exactly 30 minutes. To keep a constant volume, cover the beaker with a cooling device such as a 500 ml round-bottomed flask in which cold water is circulated. Stop boiling by adding approximately 50 ml cold water and filter immediately under vacuum through an asbestos filter previously prepared as shown in 4.8. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Wash the residue with 5 lots of approximately 100 ml of very hot water to obtain a final volume of filtrate of 800 ml. Transfer the residue quantitatively to the beaker (4.1) which has first been fitted with a porcelain disc (4.2) to regulate the boiling. Add 200 ml potassium hydroxide solution (3.4). Bring rapidly to the boil and leave to boil for exactly 30 minutes. Add approximately 50 ml cold water and filter immediately under vacuum through a fresh asbestos filter previously prepared as shown in 4.8. Wash the residue with very hot water until the washing water is neutral (test with litmus paper), then 3 times with acetone (3.6) (approximately 100 ml acetone in all). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Transfer the residue quantitatively to an ashing crucible (4.5), break up if necessary and dry to constant weight in the drying oven at 130°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Leave to cool in the desiccator (4.7) and weigh rapidly. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place the crucible in the muffle-furnace (4.6) and leave to ash for 30 minutes at 900°C. Leave to cool in the desiccator (4.7) and weigh rapidly. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank test applying the same procedure to the treated asbestos (3.2), but without the sample. Loss of weight resulting from ashing of the 6 g asbestos must not exceed 10 mg. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The crude fibre content, as a percentage of the sample, is given by the formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
where: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
0.3%, in absolute value, for crude fibre contents less than 10%; 3%, relative to the higher result, for crude fibre contents equal to or greater than 10%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Feeding stuffs containing more than 10% oil must be defatted prior to analysis with light petroleum (3.7). To do this, place the test sample (3 g weighed to the nearest mg) on an asbestos filter (4.8). Cover 3 times with approximately 50 ml light petroleum (3.7) and each time filter carefully under vacuum. Transfer the defatted test sample and the asbestos quantitatively to a beaker (4.1) and continue the analysis as shown in 5. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Feeding stuffs containing oil which cannot be extracted directly must be defatted as shown in 7.1 and defatted a further time after the acid attack has been washed from the residue. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To do this, wash the residue 3 times with acetone (3.6) (100 ml in all), then 3 times with 50 ml light petroleum (3.7). Then transfer the residue quantitatively to a beaker (4.1) and continue the analysis as shown in the second paragraph of 5 (treatment with potassium hydroxide solution). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.3 If the feeding stuffs are rich in calcium (more than 2% calcium), place the test sample (3 g, weighed to the nearest mg) in a beaker (4.1) with 100 ml hydrochloric acid 0.5 N (3.5) and leave to stand in a cool temperature for 5 minutes. Filter immediately and wash in cold water. Use as a filtration aid the 2.0 g asbestos specified for boiling with sulphuric acid. If filtration proves difficult, dilute the suspension with acetone (3.6). Then proceed as shown in 5. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
10. DETERMINATION OF BETACAROTENE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the betacarotene content of feeding stuffs. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The betacarotene is extracted with light petroleum, purified by column chromatography and measured spectrophotometrically at 450 nm. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Light petroleum, boiling range 40--60°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Light petroleum, boiling range 80--100°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Bone meal. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Standard betacarotene. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Kjeldahl flask. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Chromatography column, internal diameter approximately 25 mm. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Weigh, to the nearest mg, 1--3 g of the sample, depending on its probable betacarotene content, and place in a Kjeldahl flask (4.1). Add 50 ml of light petroleum (3.2) and heat on a steam bath for 1 hour. Cool the flask and its contents and decant the liquid into a column of bone meal previously wetted with light petroleum (3.1). Apply light suction to the column of bone meal so that the eluate drops slowly through. Rinse repeatedly the flask and the residue therein with small quantities of light petroleum (3.1). Pass each rinsing through the column and continue to elute with light petroleum (3.1) until a colorless eluate appears. Make up the total eluate obtained to 200 ml. Measure the optical density in a spectrophotometer (4.3) at 450 nm. Determine the betacarotene content by reference to a calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Calibration curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Dissolve 50 mg betacarotene (3.4) in 500 ml light petroleum (3.1) and from this stock solution prepare a series of standard solutions containing between 0.2--2.5/ug betacarotene per ml. Plot the calibration curve, using the optical density values as the ordinates and the corresponding quantities of betacarotene as the abscissae. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the betacarotene content of the sample by reference to the calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
11. DETERMINATION OF TOTAL PHOSPHORUS |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(a) VOLUMETRIC METHOD |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the total phosphorus content of feeding stuffs and mineral mixtures rich in phosphorus. For products low in phosphorus a photometric method may be used. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed and dissolved in acid. The phosphorus is precipitated as quinolinium molybdophosphate. The precipitate is dissolved in alkali the excess of which is back titrated. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Calcium oxide. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Hydrochloric acid, d: 1.18. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Nitric acid, d: 1.42. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Citric molybdate: stir 54 g of molybdic anhydride with 200 ml of water and 11 g of sodium hydroxide until the molybdic anhydride dissolves; heat the mixture to assist solution. Dissolve 60 g of citric acid in a further 300 ml of water and add 140 ml of hydrochloric acid (3.2) to it. Pour the molybdate solution into the acid solution. Stir throughout the addition. Cool the whole and, if necessary, filter through a pad of paper pulp. Dilute the filtrate with water to 1 litre and discharge the slight green or blue colour by dropwise addition of a 1% solution of potassium bromate: store the reagent in the dark. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Quinoline: dilute 60 ml of hydrochloric acid (3.2) to 400 ml with water and heat to between 70--80°C. Pour 50 ml of quinoline into the diluted acid in a thin stream stirring continuously. After the quinoline has dissolved, cool the solution, dilute to 1 litre with water, and filter through a pad of filter paper pulp. Do not wash the pulp. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Indicator solution: dissolve 0.10 g of thymol blue in 2.2 ml of 0.1 N sodium hydroxide solution, add 50 ml of ethanol (95--96% v/v) and dilute the whole to 100 ml with water and mix well. Mix 3 volumes of this solution with 1 volume of 0.1% solution of phenolphthalein in 60% ethyl alcohol. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Hydrochloric acid, 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Hydrochloric acid, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Hydrochloric acid, 25% (v/v): dilute 25 ml of hydrochloric acid (3.2) with water to 100 ml. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Sodium hydroxide, 0.5 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Sodium hydroxide, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Platinum dish. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Electric muffle-furnace with thermostat set at 500°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, 5 g of sample (if phosphorus content does not exceed 6 per cent) or 2.5 g (if phosphorus content exceeds 6 per cent) into a platinum dish (4.1). Add 1 g of calcium oxide (3.1) and mix thoroughly with a little water. Dry the mixture and incinerate at a temperature not exceeding 500°C until the bulk of the organic matter is destroyed. Cool and transfer the contents to a 250 ml beaker, add 10 ml of water and slowly add 12 ml of hydrochloric acid (3.2) taking precautions to avoid loss by effervescence and finally add 5 ml of nitric acid (3.3). Heat to incipient boiling and keep at this temperature for 10 minutes. Dilute with 100 ml of water and boil for a further 10 minutes. Filter into a 500 ml volumetric flask and wash the residue with hot water. Transfer the filter paper and residue to the original platinum dish (4.1) and incinerate until all the carbon is destroyed. Heat the ash with hydrochloric acid (3.9) for 5 minutes and add to the filtrate, cool, and dilute to 500 ml, mix well and filter. Discard the first 20 ml of the filtrate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Transfer a suitable volume (containing less than 30 mg of phosphorus and preferably about 20 mg of phosphorus) to a 500 ml stoppered conical flask and dilute to 150 ml with water. Add 50 ml of citric-molybdate solution (3.4), heat to incipient ebullition and keep at this temperature for 3 minutes, then heat to boiling point. From a burette add slowly 25 ml of quinoline solution (3.5) with constant swirling, the first few ml being added dropwise, the rest in a slow stream, maintaining gentle boiling throughout. Immerse the flask in boiling water for 5 minutes and cool to 15°C in running water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Filter the contents of the flask through a pad of filter paper pulp and wash the flask, precipitate and filter with successive portions of cold water until free from acid. Transfer the filter pad and precipitate to the original flask using not more than 100 ml of water. Stopper the flask and shake vigorously to disperse pulp and precipitate. Remove stopper, wash with water and add washings to flask. Add a measured volume of 0.5 N sodium hydroxide solution (3.10) sufficient to dissolve the precipitate and leave a few ml in excess. Shake until the precipitate dissolves and titrate the excess sodium hydroxide with 0.5 N hydrochloric acid (3.7) using indicator solution (3.6), the end point being taken as the sharp change from green-blue to yellow. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank determination on all the reagents, omitting only the sample, and using hydrochloric acid (3.8) and sodium hydroxide (3.11) for the titration. Calculate the blank in terms of 0.5 N sodium hydroxide and subtract it from the original result. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 ml of 0.5 N sodium hydroxide = 0.596 mg phosphorus. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
12 DETERMINATION OF TOTAL PHOSPHORUS |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(b) PHOTOMETRIC METHOD |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the total phosphorus content of feeding stuffs and mineral mixtures low in phosphorus. For products rich in phosphorus a volumetric method may be used. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is mineralised, either by dry combustion (in the case of organic feeding stuffs) or by acid digestion (in the case of mineral compounds and liquid feeding stuffs), and placed in an acid solution. The solution is treated with molybdovanadate reagent. The optical density of the yellow solution thus formed is measured in a spectrophotometer at 430 nm. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Calcium carbonate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Hydrochloric acid, d: 1.1 (approximately 6 N). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Nitric acid, d: 1.045. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Nitric acid, d: 1.38--1.42. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sulphuric acid, d: 1.84. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Molybdovanate reagent: mix 200 ml of ammonium heptamolybdate solution (3.6.1), 200 ml of ammonium monovanadate solution (3.6.2) and 134 ml of nitric acid (3.4) in a 1 litre graduated flask. Make up to volume with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6.1 Ammonium heptamolybdate solution: dissolve in hot water 100 g of ammonium heptamolybdate ((NH4)6 Mo7O24.4H2O). Add 10 ml of ammonia (d: 0.91) and make up to 1 litre with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6.2 Ammonium monovanadate solution: dissolve 2.35 g of ammonium monovanadate (NH4VO3) in 400 ml of hot water. Stirring constantly, slowly add 20 ml of dilute nitric acid (7 ml of HNO3 (3.4) + 13 ml of H2O) and make up to 1 litre with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Standard solution of 1 mg phosphorus per ml: dissolve 4.387 g of potassium dihydrogen phosphate (KH2PO4) in water. Make up to 1 litre with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Silica or porcelain ashing crucibles. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Electric muffle-furnace with thermostat set at 550°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Kjeldahl flask. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Graduated flasks and precision pipettes. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Test tubes with stoppers. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the solution |
|||||||||||||||||||||||||||||||||||||||||||||||
|
According to the nature of the sample, prepare a solution as indicated in 5.1.1 or 5.1.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.1 Usual procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, 1 g or more of the sample. Place the test sample in a Kjeldahl flask, add 20 ml of sulphuric acid (3.5), shake to impregnate the substance completely with acid and to prevent it from sticking to the sides of the flask, heat and keep at boiling point for 10 minutes. Leave to cool slightly, add 2 ml of nitric acid (3.4), heat gently, leave to cool slightly, add a little more nitric acid (3.4) and bring back to boiling point. Repeat this procedure until a colourless solution is obtained. Cool, add a little water, decant the liquid into a 500 ml graduated flask, rinsing the Kjeldhal flask with hot water. Leave to cool, make up to volume with water, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.2 Samples containing organic substances and free from calcium and magnesium dihydrogen phosphates |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, about 2.5 g of the sample in an ashing crucible (4.1). Mix the test sample until completely merged with 1 g of calcium carbonate (3.1). Ash in the oven at 550°C ± 5°C until white or grey ash is obtained (a little charcoal does not matter). Transfer the ash into a beaker. Add 20 ml of water and hydrochloric acid (3.2) until effervescence ceases. Add a further 10 ml of hydrochloric acid (3.2). Place the beaker on a sand bath and evaporate until dry to make the silica insoluble. Redissolve the residue in 10 ml of nitric acid (3.3) and boil on the sand bath for 5 minutes. Decant the liquid into a 500 ml graduated flask, rinsing the beaker several times with hot water. Leave to cool, make up to volume with water, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Development of colouration and measurement of optical density |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Dilute an aliquot part of the filtrate obtained by 5.1.1 or 5.1.2 to obtain a phosphorus concentration of not more than 40µg/ml. Place 10 ml of this solution in a test tube (4.6) and add 10 ml of molybdovanadate reagent (3.6). Mix and leave to stand for at least 10 minutes at 20°C. Measure the optical density in a spectrophotometer (4.5), at 430 nm against a solution obtained by adding 10 ml of the molybdovanadate reagent (3.6) to 10 ml of water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Calibration curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
From the standard solution (3.7) prepare solutions containing respectively 5, 10, 20, 30 and 40 g of phosphorus per ml. Take 10 ml of each of these solutions and add thereto 10 ml of molybdovanadate reagent (3.6). Mix and leave to stand for at least 10 minutes at 20°C. Measure the optical density as indicated in 5.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Trace the calibration curve by plotting the optical densities against the corresponding quantities of phosphorus. For concentrations between 0--40µg/ml the curve will be linear. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Determine the amount of phosphorus in the test sample by using the calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample should not exceed: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3% relative to the higher result, for phosphorus contents of less than 5%; |
|||||||||||||||||||||||||||||||||||||||||||||||
|
0.15% in absolute value, for phosphorus contents equal to or greater than 5%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
13. DETERMINATION OF CRUDE ASH |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the crude ash content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed at 550°C; the residue is weighed. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagent |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Ammonium nitrate solution, 20% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Hot plate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Electric muffle-furnace with thermostat. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Crucibles for ashing made of platinum or an alloy of platinum and gold, or silica or porcelain. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample (2.5 g in the case of products which have a tendency to swell) and place in a crucible for ashing (4.3) which has first been heated to 550°C ± 5°C cooled in a "desiccator" and weighed. Place the crucible on the hot plate (4.1) and heat gradually until the substance carbonises. Put the crucible into the muffle-furnace (4.2) set at 550°C ± 5°C. Keep at this temperature until white, light grey or reddish ash is obtained which appears to be free from carbonaceous particles. Place the crucible in a desiccator, leave to cool and weigh immediately. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the weight of the residue and express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Substances which are difficult to ash must be subjected to an initial ashing of at least 3 hours, cooled and then a few drops of 20% solution of ammoniun nitrate (3) added to it (carefully, to avoid dispersal of the ash or the formation of lumps). Continue ashing after drying in the oven. Repeat the operation as necessary until ashing is complete. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 In the case of substances resistant to the treatment described under 7.1, proceed as follows: after ashing for 3 hours, place the ash in warm water and filter through a small, ash-free filter. Ash the filter and its contents in the original crucible. Place the filtrate in the cooled crucible, evaporate until dry, ash and weigh. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.3 In the case of oils and fats, weigh accurately a sample of approximately 25 g in a suitably sized crucible. Carbonise by setting light to the substance with a strip of ash-free filter paper. After combustion, moisten with as little water as possible. Dry and ash as described under 5. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
14. DETERMINATION OF ASH WHICH IS INSOLUBLE IN HYDROCHLORIC ACID |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content, in feeding stuffs, and mineral mixtures, of mineral substances which are insoluble in hydrochloric acid. Either of two methods can be used, depending on the nature of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.1 Method A: applicable to straight organic feeding stuffs and to compound feeding stuffs other than those specified under Method B. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1.2 Method B: applicable to mineral compounds and mixtures and to compound feeding stuffs whose content of substances insoluble in hydrochloric acid, as determined by Method A, is greater than 1%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2.1 Method A: the sample is ashed, the ash boiled in hydrochloric acid and the insoluble residue filtered and weighed. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2.2 Method B: the sample is treated with hydrochloric acid. The solution is filtered, the residue ashed and the ash thus obtained treated in accordance with Method A. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Hydrochloric acid solution, 3 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Trichloroacetic acid solution, 20% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Trichloroacetic acid solution, 1% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Hot plate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Electric muffle-furnace with thermostat. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Crucibles for ashing made of platinum or an alloy of platinum and gold, or of silica or porcelain. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Method A: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Ash the sample using the method prescribed in this schedule for the determination of crude ash. Ash obtained from that analysis may also be used. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place the ash in a 250--400 ml beaker using 75 ml of hydrochloric acid 3 N (3.1). Bring slowly to the boil and boil gently for 15 minutes. Filter the warm solution through an ash-free filter paper and wash the residue with warm water until the acid reaction is no longer visible. Dry the filter containing the residue and ash in a tarred crucible at a temperature of not less than 550°C and not more than 700°C. Cool in a desiccator and weigh. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Method B: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample, and place in a 250--400 ml beaker. Add 25 ml of water and 25 ml of hydrochloric acid 3 N (3.1) successively, mix and wait for effervescence to cease. Add a further 50 ml of hydrochloric acid 3 N (3.1). Wait for any release of gas to cease then place the beaker in a boiling water bath and keep it there for 30 minutes (or longer if necessary) in order to hydrolyse thoroughly any starch which may be present. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Filter while warm through an ash-free filter and wash the filter in 50 ml of warm water (see observation, 7). Place the filter containing the residue in a crucible for ashing (4.3), dry and ash at a temperature of not less than 550°C and not more than 700°C. Place the ash in a 250--400 ml beaker using 75 ml of hydrochloric acid 3 N (3.1); continue as described in the second sub-paragraph of 5.1. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the weight of the residue and express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observation |
|||||||||||||||||||||||||||||||||||||||||||||||
|
If filtration proves difficult recommence the analysis, replacing the 50 ml of hydrochloric acid 3 N (3.1) by 50 ml of 20% trichloroacetic acid (3.2) and washing the filter in a warm solution of 1% trichloroacetic acid (3.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
15. DETERMINATION OF CALCIUM. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the total calcium content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed, the ash treated with hydrochloric acid and the calcium precipitated as calcium oxalate. The precipitate is dissolved in sulphuric acid and the oxalic acid formed is titrated with a solution of potassium permanganate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Hydrochloric acid, d: 1. 14. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Nitric acid, d: 1.40. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Sulphuric acid, d: 1.13. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Ammonia, d: 0.98. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Cold saturated solution of ammonium oxalate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Citric acid solution, 30% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Ammonium chloride solution, 5% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Bromocresol green solution, 0.04% (w/v.) |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Potassium permanganate solution, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Electric muffle-furnace with air circulation and thermostat. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Platinum, silica or porcelain crucibles for ashing. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Glass filter crucibles of G4 porosity. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample (or more if necessary), incinerate at 550°C and transfer the ash to a 250 ml beaker. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Add 40 ml of hydrochloric acid (3.1), 60 ml of water and a few drops of nitric acid (3.2). Bring to the boil and keep at boiling point for 30 minutes. Cool and transfer the solution to a 250 ml volumetric flask. Rince, bring the volume up to the mark with water, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Using a pipette, transfer to a 250 ml beaker an aliquot containing 10-40 mg of calcium according to the assumed calcium content. Add 1 ml of citric acid solution (3.6) and 5 ml of ammonium chloride solution (3.7). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Make the volume up to approximately 100 ml with water. Bring to the boil, add eight to ten drops of bromocresol green solution (3.8) and 30 ml of a warm solution of ammonium oxalate (3.5). If a precipitate forms, dissolve it by adding a few drops of hydrochloric acid (3.1). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Neutralise very slowly with ammonia (3.4), stirring continuously until a pH reading of 4.4-4.6 is obtained (i.e. when the indicator changes colour). Place the beaker in a boiling water bath and keep there for 30 minutes to allow the precipitate which has formed to settle. Remove the beaker from the water bath. Leave it to stand for an hour and filter through a G4 filter crucible (4.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Wash the beaker and the crucible with water until the excess ammonium oxalate is completely removed (the absence of chloride in the washing water indicates that they have been sufficiently washed). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Dissolve the precipitate on the filter in 50 ml of warm sulphuric acid (3.3). Rinse the crucible with warm water and make the filtrate up to approximately 100 ml. Bring the temperature up to 70-80°C and titrate drop by drop with a solution of potassium permanganate (3.9) until a pink colour is obtained which lasts for 1 minute. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1 ml of potassium permanganate 0.1 N corresponds to 2.004 mg of calcium. Express the result obtained as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 For very low contents of calcium proceed as follows: filter the calcium oxalate precipitate through an ash-free filter paper. After washing, dry the filter and ash at 550°C in a platinum crucible. Redissolve the residue in a few drops of sulphuric acid (3.3), evaporate until dry, incinerate again at 550°C and weigh. If W is the weight of the calcium sulphate obtained, the calcium content of the aliquot amount taken as a sample = W, 0.2944. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 If the sample consists solely of mineral substances, dissolve in hydrochloric acid (3.1) without ashing it first. In the case of products such as calcium aluminium phosphate which are difficult to dissolve in acid, melt as follows by an alkaline process before dissolving: mix the sample to be analysed thoroughly in a platinum crucible with a mixture five times its weight, consisting of equal amounts of potassium carbonate and sodium carbonate. Heat carefully until the mixture is completely melted. Cool, and dissolve in hydrochloric acid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.3 If the magnesium content of the sample is high, precipitate the calcium oxalate a second time. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
16 DETERMINATION OF IODINE |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the iodine content of feeding stuffs and mineral mixtures (other than those containing little or no organic matter). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed, extracted with water and the liberated iodine titrated with sodium thiosulphate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Absolute ethanol. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Anhydrous sodium carbonate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Bromine water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Methyl Orange indicator solution. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Orthophosphoric acid solution, 85% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Potassium iodide. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Salicylic acid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Sodium hydroxide solution, 50% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Sodium thiosulphate solution, 0.005 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Nickel crucible. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Muffle-furnace with thermostat set at 500°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Place a weighed portion of the sample containing 3-4 mg of iodine in a 200-300 ml nickel crucible (4.1). Add about 5 g of anhydrous sodium carbonate (3.2), 5 ml of sodium hydroxide solution (3.8) and 10 ml of ethanol (3.1). Ensure that the entire sample is moist. Dry the dish and its contents at 100°C for about 30 minutes, then heat in muffle-furnace (4.2) to 500°C. Maintain temperature at 500°C ± 5°C for 15 minutes. Then cool dish and contents. Add 25 ml of water, cover dish with watch glass and boil gently for 10 minutes. Transfer contents of dish to filter paper 18 cm in diameter. Wash dish and paper with boiling water into a beaker until the filtrate is approximately 300 ml. Neutralise the filtrate with 85 per cent orthophosphoric acid (3.5) using methyl orange indicator (3.4). Add 1 ml of acid in excess. Add excess bromine water (3.3) and boil solution until colourless and for a further 5 minutes thereafter. Add a few crystals of salicylic acid (3.7) and cool solution to about 20°C. Add 1 ml of orthophosphoric acid (85 per cent) (3.5) and about 0.5 g of potassium iodide (3.6) and titrate the solution with 0.005 N sodium thiosulphate (3.9). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
17 DETERMINATION OF WATER-SOLUBLE CHLORIDES |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of water-soluble chlorides, expressed as sodium chloride, in all feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The chlorides are dissolved in water. If the product contains organic matter it is clarified. The solution is slightly acidified with nitric acid and the chlorides precipitated in the form of silver chloride by means of a solution of silver nitrate. The excess silver nitrate is titrated with a solution of ammonium thiocyanate, by Volhard's method. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Ammonium thiocyanate solution, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Silver nitrate solution, 0.1 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Ammonium ferric sulphate, saturated solution. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Nitric acid, d: 1.38. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Diethyl ether or light petroleum, boiling range 40-60°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Acetone. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Carrez I solution: dissolve in water 21.9 g of zinc acetate (Zn (CH3COO2). 2H2O) and 3 g of glacial acetic acid. Make up to 100 ml with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Carrez II solution: dissolve in water 10.6 g of potassium ferrocyanide, K4[Fe (CN)6] 3H2O. Make up to 100 ml with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Active carbon, free from chlorides and not absorbing them. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Mixer (tumbler): approximately 35-40 rpm. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the solution |
|||||||||||||||||||||||||||||||||||||||||||||||
|
According to the nature of the sample, prepare a solution as shown under 5.1.1, 5.1.2 or 5.1.3. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
At the same time carry out a blank test omitting the sample to be analysed. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.1 Samples free from organic matter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, a sample of not more than 10 g and containing not more than 3 g of chlorine in the form of chlorides. Place with 400 ml of water in a 500 ml volumetric flask at approximately 20°C. Mix for 30 minutes in the tumbler, bring up to volume, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.2 Samples containing organic matter, excluding the products listed under 5.1.3. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample and place with 1 g of active carbon (3.9) in a 500 ml volumetric flask. Add 400 ml of water at approximately 20°C and 5 ml of Carrez solution I (3.7), stir, then add 5 ml of Carrez solution II (3.8). Mix for 30 minutes in the tumbler, bring up to volume, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.3 Cooked feeding stuffs, linseed cakes and meal, products rich in linseed meal and other products rich in mucilage or in colloidal substances (for example, dextrinated starch). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Prepare the solution as described under 5.1.2 but do not filter. Decant (if necessary centrifuge), remove 100 ml of the supernatant liquid and transfer to a 200 ml measuring flask. Mix with acetone (3.6) and bring up to volume with this solvent, mix and filter. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Titration |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Using a pipette, transfer to an Erlenmeyer flask from 25-100 ml of the filtrate (according to the assumed chlorine content) obtained as described under 5.1.1, 5.1.2 or 5.1.3. The aliquot portion must not contain more than 150 mg of chlorine (Cl). Dilute if necessary to not less than 50 ml with water, add 5 ml of nitric acid (3.4), 20 ml of saturated solution of ammonium ferric sulphate (3.3) and two drops of ammonium thiocyanate solution (3.1) transferred by means of a burette filled up to zero mark. Using a burette, transfer the silver nitrate solution (3.2) in such a way that an excess of 5 ml is obtained. Add 5 ml of diethyl ether or light petroleum (3.5) and shake hard to coagulate the precipitate. Titrate the excess silver nitrate with the ammonium thiocyanate solution (3.1) until the reddish-brown tint has lasted for 1 minute. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The amount of chlorine (W), expressed as sodium chloride, present in the volume of filtrate taken for titration is calculated by using the following formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
W = 5.845 (V1 - V2) mg |
|||||||||||||||||||||||||||||||||||||||||||||||
|
where: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
If the blank test indicates that silver nitrate solution 0.1 has been consumed deduct this value from the volume (V1- V2). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Titration may also be carried out by potentiometry. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.2 In the case of products which are very rich in oils, first defat with diethyl ether or light petroleum. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7.3 In the case of fish meal, titration may be carried out by Mohr's method. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
18. DETERMINATION OF COBALT. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the cobalt content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed, dissolved in acid and a colour complex formed between the cobalt and nitroso-R salt. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Cobalt sulphate solution: dissolve 0.2385 g of cobalt sulphate in water and dilute to 1 litre (1 ml of this solution is equal to 0.05 mg of cobalt). Dilute the solution to a suitable concentration to prepare standard curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Hydrated sodium acetate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Hydrochloric acid, d: 1.18. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Hydrochloric acid solution, 50% (v/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Nitric acid, d: 1.42. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Nitric acid solution, 10% (v/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Nitroso-R salt (0.2% aqueous solution). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Ashing crucibles. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Muffle-furnace with thermostat set at 500°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Incinerate a weighed sample at 500°C ± 5°C in order to destroy the organic matter. Transfer the ash to a 100 ml volumetric flask and boil with 10 ml hydrochloric acid (3.3) and 25 ml water for 5 minutes, cool, dilute to volume and allow to settle. Evaporate an aliquot (containing not more than 0.5 mg cobalt; if the copper content of the aliquot exceeds 10 mg it must be removed with hydrogen sulphide) of the clear solution almost to dryness, add 1 ml nitric acid (3.5) and continue to evaporate just to dryness. Dissolve the residue in 10 ml water containing 0.5 ml hydrochloric acid (3.4) and 0.5 ml nitric acid (3.6). Boil for a few minutes to dissolve any solid material. Add 2.5 ml of a 0.2 per cent aqueous solution of nitroso-R salt (3.7) and 2 g hydrated sodium acetate (3.2). The pH of the solution should be close to 5.5; check with bromocresol green indicator or a pH meter. Boil for 1 minute, add 1 ml hydrochloric acid (3.3) and boil again for 1 minute. Cool to room temperature and dilute to 25 ml in a volumetric flask (filter if necessary). Determine the optical density in a spectrophotometer at 540 nm (4.2). The colour should be read within 2 hours. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Preparation of standard curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To 1, 2, 3 etc, ml of cobalt sulphate solution (3.1) add 40 ml of water containing 2.0 ml of hydrochloric acid (3.4); 2.0 ml of nitric acid (3.6); 10.0 ml of nitroso-R salt (3.7) and 8 g of hydrated sodium acetate (3.2). Similarly prepare a blank but omitting cobalt sulphate solution and nitroso-R salt. Heat standard and blank to boiling and add 4 ml of hydrochloric acid (3.3): boil for 1 minute, cool and dilute to 100 ml in a volumetric flask. Plot a standard curve after determining the optical densities in a spectrophotometer at 540 nm (4.2). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the cobalt content of the sample by reference to the calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
19. DETERMINATION OF COPPER-DIETHYLDITHIOCARBAMATE SPECTROPHOTOMETRIC METHOD |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the copper content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed and the residue treated with hydrochloric acid. Copper is extracted from the resulting solution as its diethyldithiocarbamate complex into carbon tetrachloride. The copper content is measured at 436 nm, by reference to a calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The water used should be free from copper, |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Carbon tetrachloride, redistilled. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Sodium diethyldithiocarbamate solution: dissolve 1 g sodium diethyldithiocarbamate in water and dilute to 100 ml. Filter the solution if it is not clear. The solution may be stored, protected from light, in a refrigerator but it should not be used after seven days. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 EDTA-citrate solution: dissolve 20 g ammonium citrate and 5 g of the disodium salt of ethylenediaminetetra-acetic acid (EDTA) in water and dilute to 100 ml. To purify, add 0.1 ml sodium diethyldithiocarbamate solution (3.2) and extract with carbon tetrachloride (3.1). Add a further quantity of sodium diethyldithiocarbamate solution (3.2) to ensure that it is in excess. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Ammonium hydroxide solution approximately 6 N: this may be prepared by passing gaseous ammonia into water, or by purifying ammonia solution as described for the EDTA-citrate solution (3.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sulphuric acid solution, 2 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Hydrochloric acid solution, 50% (v/v): dilute an appropriate volume of hydrochloric acid (density 1.18 g/ml) with an equal volume of water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Hydrochloric acid solution, 2 N. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Nitric acid solution, 30% (v/v): dilute 30 ml of nitric acid (d: 1.42 g/ml) with water to 100 ml. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Copper standard solution: weigh to the nearest 0.1 mg, 393 mg of copper sulphate (CuSO4. 5H2O), dissolve in 100 ml 2 N sulphuric acid (3.5) and dilute to one litre with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Copper standard working solution: dilute 5 ml of the copper sulphate standard solution (3.9) to 250 ml with 2 N sulphuric acid (3.5) immediately before use. 1 ml of this solution = 2µg copper (Cu). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Thymol blue indicator solution: dissolve 0.1 g thymol blue in 2.15 ml of 0.1 N sodium hydroxide and dilute to 100 ml with water. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Spectrophotometer with 10 mm cells. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Preparation of the test sample |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Grind the sample to pass through a stainless steel sieve having apertures about, 1 mm square. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6.1 Dissolution of sample |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 0.001 g, approximately 10 g of the sample prepared under 5 into a silica dish or basin, and place a silica cover on top. Transfer to a cool muffle-furnace. Raise the temperature to 450° ± 10°C and allow to ash until all the carbonaceous matter has disappeared; a slow current of air through the furnace during the initial stages of ashing is desirable. In the case of high-fat content materials, care must be taken to avoid ignition of the sample. When all the organic matter has been destroyed, cool, add 10 ml 50% (v/v) hydrochloric acid solution (3.6) and evaporate to dryness on a water-bath. Extract the soluble salts from the residue with two successive 10 ml portions of boiling 2 N hydrochloric acid solution (3.7) decanting the solution each time through the same suitable filter-paper (1) into a 50 ml graduated flask. Then add 5 ml of 50% (v/v) hydrochloric acid solution (3.6) and about 5 ml of 30% (v/v) nitric acid solution (3.8) to the residue in the basin, and evaporate the mixture to dryness on a hot-plate at low heat. Finally, add a further 10 ml of boiling 2 N hydrochloric acid solution (3.7) to the residue and filter the solution through the same filter-paper into the flask. Wash the basin and the filter with water, and collect the washings in the flask, make up to the mark with water and mix. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(1) Whatman No. 541 or equivalent. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6.2 Determination |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Transfer to a separating funnel a suitable aliquot of the solution prepared in accordance with 6.1 (or a dilution of this solution in N hydrochloric acid), containing not more than 50µg of copper. Add 10 ml EDTA-citrate solution (3.3), two drops of thymol blue indicator solution (3.11) and ammonium hydroxide solution (3.4) until the mixture is coloured green or bluish-green. Cool the mixture, add 1 ml of sodium diethyldithiocarbamate solution (3.2) and, from a burette, 15 ml of carbon tetrachloride (3.1). Stopper the funnel, shake vigorously for two minutes and allow the layers to separate. Place a piece of cotton-wool in the stem of the funnel and run off the carbon tetrachloride layer into a dry 10 mm spectrophotometer cell (4.1). Avoid undue exposure of the solution to light. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Measure immediately the absorbance of the sample solution at 436 nm, against carbon tetrachloride as reference. Determine the quantity of copper by reference to the calibration curve (6.4). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6.3 Blank test |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank test omitting only the sample and following the procedure described under 6.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6.4 Calibration curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To a series of separating funnels transfer 10 ml EDTA-citrate solution (3.3) and the following amounts of copper standard working solution (3.10) and 2 N sulphuric acid (3.5): |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Proceed as for the test solution, as described in 6.2 commencing " . . . two drops thymol blue indicator (3.11) . . .". Measure the absorbances of the solutions and plot the calibration curve using absorbances as the ordinates and the corresponding quantities of copper in µg as the abscissae. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
7. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The copper content in mg/kg of sample is given by the formula: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
20. DETERMINATION OF IRON. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the iron content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed, dissolved in aqueous citric acid and complexed with thioglycollic acid in the presence of ammonia. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Ammonia solution, 10% (v/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Citric acid solution, 20% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Thioglycollic acid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Citric acid. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Ashing crucibles. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Muffle-furnace with thermostat set at 500°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Analysis of sample |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, a portion of sample containing about 1 mg iron and ash at 500°C ± 5°C. Transfer the residue to a 100 ml volumetric flask and add 4 g of citric acid (3.4) and 80 ml of water. Mix well and allow to stand for about 1 hour. Dilute the mixture to volume and Filter. Transfer a 5 ml aliquot of the solution so obtained to a marked colorimeter tube and add 3 drops of thioglycollic acid (3.3), followed by ammonia solution (3.1) drop-wise until a stable reddish colour develops (care being taken to avoid adding excess ammonia). Dilute this solution to 10 ml with water and determine the optical density in a spectrophotometer at 540 nm (4.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Preparation of a standard curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Prepare a solution containing 50µg/ml of iron and transfer the following quantities to colorimeter tubes marked at 10 ml Í 0.4, 0.8, 1.2, 1.6 and 2.0 ml, giving 20, 40, 60, 80 and 100µg of iron respectively. Add to each tube 1 ml of citric acid solution (3.2) and sufficient water to bring the volume to approximately 5 ml. Prepare a blank by adding to another tube 1 ml of citric acid solution (3.2) and 4 ml of water. Add to each tube 3 drops of thioglycollic acid (3.3) mix thoroughly and add sufficient quantity (about 4 ml) of ammonia solution (3.1) as will suffice to produce a stable reddish colour. Dilute the contents of each tube to volume with water and mix thoroughly. Plot a standard curve from these solutions using a colorimeter or spectrophotometer at 540 nm (4.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
From calibration curve calculate the iron content of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
21. DETERMINATION OF MANGANESE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the manganese content of feeding stuffs and mineral mixtures. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed and the manganese is oxidised to permanganate by means of periodate in acid solution, and the permanganate is then determined spectrophotometrically. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Hydrochloric acid, d: 1.18. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Nitric acid, d: 1.42. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Orthophosphoric acid, 85% (w/v). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Potassium periodate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Potassium permanganate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Sodium oxalate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Preparation of standard solution: dissolve by boiling with water 1.4383 g of potassium permanganate, dilute to 1 litre and allow to stand for several days. Filter the solution through an asbestos pad on a Gooch crucible and standardise with sodium oxalate. The solution so obtained shall contain at least 500 mg/litre of manganese. Transfer to a beaker an aliquot part of the solution containing 20 ml of manganese. Add 100 ml of water, 15 ml of orthophosphoric acid (3.3) and 0.3 g of potassium periodate (3.4). Bring solution to boiling point, cool and dilute to 1 litre. Protect the solution from light. Dilute the solution (containing 20 mg/litre of manganese) with water which has been boiled with 0.3 g of potassium periodate per litre to make convenient working standards of known concentrations approximately the same as those to be compared. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Ashing crucible. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Muffle-furnace with thermostat set at 500°C. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Analysis of sample |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, 5-15g of sample and ash at 500°C 5°C. Place a suitable quantity of ash in a 250 ml beaker, add 15 ml water and cover with a watch glass. Slowly add 15 ml hydrochloric acid (3.1) and 3 ml nitric acid (3.2). Heat to boiling and boil gently until the soluble material is in solution. Dilute with 15-25 ml water, boil again gently for a few minutes and cool; wash the watch glass, transfer the solution and washings to a 100 ml volumetric flask and make up to volume; mix thoroughly and stand overnight. Pipette 50 ml of the solution into an evaporating basin and evaporate to dryness on a steam bath. Add nitric acid (3.2) and again evaporate just to dryness. Again add nitric acid and evaporate just to dryness to eliminate chlorine completely. Dissolve the residue in 25-40 ml water and add minimum quantity of nitric acid (3.2); then add 1 ml orthophosphoric acid (3.3) and heat on a steam bath; add 0.3 g potassium periodate (3.4) and continue heating (at 90-100°C) until maximum colour development takes place (30 minutes approximately). Cool and dilute to 100 ml. Determine the optical density in a spectrophotometer at 530 nm (4.3). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Calibration curve |
|||||||||||||||||||||||||||||||||||||||||||||||
|
From standard solution (3.7) prepare a series of reference solutions of known concentrations (containing between 1-10µg of manganese per ml) and from these construct a calibration curve. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
From the calibration curve calculate the manganese content of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
22. DETERMINATION OF MAGNESIUM. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
by atomic obsorption spectrophotometry. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
|||||||||||||||||||||||||||||||||||||||||||||||
|
To determine the magnesium content of feeding stuffs. It is particularly appropriate for determining magnesium contents lower than 5%. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The sample is ashed and dissolved in dilute hydrochloric acid. If it contains no organic substances, it is dissolved directly in dilute hydrochloric acid. The solution is diluted and the magnesium content determined by atomic absorption spectrophotometry at 285.2 nm, by comparison with standard solutions. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Hydrochloric acid, d: 1.16 |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Hydrochloric acid, d: 1.19. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Magnesium ribbon or wire, or magnesium sulphate heptahydrate, dried at room temperature. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Strontium salt solution (chloride or nitrate) at 2.5% (w/v) strontium (= 76.08 g Sr Cl2 6H2O or 60.38 g Sr (No3)2 per 1000 ml.). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Standard magnesium solution: weigh, to the nearest mg, 1 g magnesium (3.3) which has previously had its oxide coating carefully removed, or the corresponding quantity (10.143 g) of magnesium sulphate heptahydrate (3.3). Place in a 1000 ml graduated flask, add 80 ml hydrochloric acid (3.1), leave to dissolve and make up to 100 ml with water. 1 ml of this solution contains 1.000 mg magnesium. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Platinum, silica or porcelain ashing crucibles. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Thermostatically controlled electric muffle-furnace. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Atomic absorption spectrophotometer. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the sample solution |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.1 Feeding stuffs composed exclusively of mineral substances. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample into a 500 ml graduated flask with 250-300 ml water. Add 40 ml hydrochloric acid (3.1), bring to the boil and keep the liquid gently boiling for 30 minutes. Leave to cool, make up to volume with water, mix and filter into a dry beaker through a dry pleated filter. Discard the first 30 ml of the filtrate. In the presence of silica, treat 5 g of sample with a sufficient quantity (15-30 ml) of hydrochloric acid (3.2), evaporate to dryness on a water bath and transfer to an oven at 105°C for 1 hour. Proceed as from the third sentence of 5.1.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.2 Feeding stuffs composed predominantly of mineral substances. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample into a crucible (4.1) and ash at 550°C in the muffle-furnace (4.2) until an ash which is free from carbonaceous particles is obtained, and leave to cool. In order to eliminate silica, add to the ash a sufficient quantity (15-30 ml) of hydrochloric acid (3.2), evaporate to dryness on a water bath and transfer to an oven at 105°C for 1 hour. Treat the residue with 10 ml hydrochloric acid (3.1) and transfer to a 500 ml graduated flask using warm water. Leave to cool and make up to volume with water. Mix and filter into a dry beaker through a dry pleated filter. Discard the first 30 ml of the filtrate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.1.3 Feeding stuffs composed predominantly of organic substances. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 5 g of the sample into a crucible (4.1) and ash at 550°C in the muffle-furnace (4.2) until an ash which is free from carbonaceous particles is obtained. Treat the ash with 5 ml hydrochloric acid (3.2), evaporate to dryness on a water bath and then dry for 1 hour in the oven at 105°C in order to render the silica insoluble. Treat the ash with 5 ml hydrochloric acid (3.1), transfer to a 250 ml graduated flask using warm water, bring to the boil, leave to cool and make up to volume with water. Mix and filter into a dry beaker through a dry pleated filter. Discard the first 30 ml of the filtrate. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Measurement by atomic absorption |
|||||||||||||||||||||||||||||||||||||||||||||||
|
By diluting the standard solution (3.5) with water, prepare at least 5 reference solutions of increasing concentration, corresponding to the optimal measuring range of the spectrophotometer (4.3). Add to each solution 10 ml strontium salt solution (3.4) and then make up the volume to 100 ml with water. Dilute with water one aliquot of the filtrate obtained from 5.1.1, 5.1.2 or 5.1.3, so as to obtain a magnesium concentration which is within the limits of concentration of the reference solutions. The hydrochloric acid concentration of this solution must not exceed 0.4 N. Add 10 ml strontium salt solution (3.4) and then make up the volume to 100 ml with water. Measure the absorption of the solution to be determined and of the reference solutions at 285.2 nm. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the quantity of magnesium in the sample by relation to the reference solutions. Express the result as a percentage of the sample. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
|||||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed 5%, relative to the higher result. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
PART II. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Form F.F.2. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Ref. No. ....................................... |
|||||||||||||||||||||||||||||||||||||||||||||||
|
CERTIFICATE OF RESULT OF ANALYSIS OF A FEEDING STUFF OR A COMPOUND FEEDING STUFF (OTHER THAN A MINERAL MIXTURE). |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
This is to certify that the above-mentioned sample, which was duly fastened and sealed, has been analysed under the direction of the *State Chemist/* Assistant State Chemist and that the result of the analysis is as follows:-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
This certificate is given under section 3*/8* of the Fertilisers, Feeding Stuffs and Mineral Mixtures Act, 1955. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
NOTE On comparison of the result of the analysis with the particulars accompanying the sample it appears that the particulars furnished-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* are correct, subject to the prescribed limits of error. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* are not correct subject to the prescribed limits of error, in the following respects:-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* Delete if inapplicable. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(1) Guaranteed contents to which Section 2 (8) of the Act applies. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
PART III |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Form F.F.3. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Ref. No. ........................................... |
|||||||||||||||||||||||||||||||||||||||||||||||
|
CERTIFICATE OF RESULT OF ANALYSIS OF A MINERAL MIXTURE |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
This is to certify that the above-mentioned sample, which was duly fastened and sealed, has been analysed under the direction of the *State Chemist/*Assistant State Chemist and that the result of the analysis is as follows:-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
This certificate is given under section *3/*8 of the Fertilisers, Feeding Stuffs and Mineral Mixtures Act, 1955. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Note: On comparison of the result of the analysis with the particulars accompanying the sample it appears that the particulars furnished-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* are correct, subject to the prescribed limits of error. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* are not correct subject to the prescribed limits of error, in the following respects:-- |
|||||||||||||||||||||||||||||||||||||||||||||||
|
* Delete if inapplicable. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
(1) Guaranteed contents to which section 2 (8) of the Act applies. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Given under my Official Seal this 1st day of September, 1978. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
JAMES GIBBONS, |
|||||||||||||||||||||||||||||||||||||||||||||||
|
Minister for Agriculture. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
EXPLANATORY NOTE. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
These Regulations amend (a) the provisions of Article 10 of the Fertilisers, Feeding Stuffs and Mineral Mixtures Regulations, 1957 ( S.I. No. 264 of 1957 ) so as to give statutory effect to certain of the provisions of Commission Directives 7½50/EEC, 71/393/EEC, 72/199/EEC, and 7¾6/EEC adopted pursuant to Council Directive 70/373/EEC of the 20th July, 1970 and (b) the forms of certificates of the results of analyses. |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||