S.I. No. 250/1978 -- European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978.
|
S.I. No. 250/1978: EUROPEAN COMMUNITIES (FEEDING STUFFS) (METHODS OF ANALYSIS) REGULATIONS, 1978. |
||||||||||||||||||||||||||||||||||||||||||||
|
EUROPEAN COMMUNITIES (FEEDING STUFFS) (METHODS OF ANALYSIS) REGULATIONS, 1978. |
||||||||||||||||||||||||||||||||||||||||||||
|
I, JAMES GIBBONS, Minister for Agriculture, in exercise of the power conferred on me by section 3 of the European Communities Act, 1972 (No. 27 of 1972), for the purpose of giving effect to Council Directive No. 70/373/EEC of 20 July, 1970, and subsequent Commission Directives supplementary thereto, hereby make the following regulations: |
||||||||||||||||||||||||||||||||||||||||||||
|
PART 1. |
||||||||||||||||||||||||||||||||||||||||||||
|
1. These Regulations may be cited as the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978, and shall come into operation on the 1st day of October, 1978. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. The methods of analysis specified in Part II of these Regulations shall be the methods of analysis to be used for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 ( S.I. No. 302 of 1974 ), and the European Communities (Feeding Stuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1977 ( S.I. No. 246 of 1977 ). |
||||||||||||||||||||||||||||||||||||||||||||
|
3. The result of an analysis of feeding stuffs carried out for the purposes mentioned in paragraph 2 of these Regulations shall be set out in a certificate in the form set out in the Schedule to these Regulations. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. (1) In any prosecution for an offence under either of the Regulations mentioned in paragraph 2 of these Regulations, the production of a certificate in the form set out in the Schedule to these Regulations, purporting to be signed by the State Chemist or the Assistant State Chemist, shall be evidence of the facts stated in the certificate and of the analysis referred to in the certificate having been carried out, unless the defendant requires the person who made the analysis to be called as a witness. |
||||||||||||||||||||||||||||||||||||||||||||
|
(2) In this paragraph-- |
||||||||||||||||||||||||||||||||||||||||||||
|
"the State Chemist" means the head of the State Laboratory or a person authorised by him for the purposes of these Regulations. |
||||||||||||||||||||||||||||||||||||||||||||
|
"the Assistant State Chemist" means the Assistant State Chemist of the State Laboratory. |
||||||||||||||||||||||||||||||||||||||||||||
|
PART II. METHODS OF ANALYSIS OF THE COMPONENTS OF FEEDINGSTUFFS AND PREMIXTURES |
||||||||||||||||||||||||||||||||||||||||||||
|
The main divisions in this Part of these Regulations are as follows:-- |
||||||||||||||||||||||||||||||||||||||||||||
|
1. General Provisions |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Determination of volatile nitrogenous bases |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Detection and identification of antibiotics of the tetracycline group |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
5. Determination of oleandomycin |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Determination of tylosin |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination of virginiamycin |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Determination of amprolium |
||||||||||||||||||||||||||||||||||||||||||||
|
9. Determination of ethopabate |
||||||||||||||||||||||||||||||||||||||||||||
|
10. Determination of dinitolmide (DOT) |
||||||||||||||||||||||||||||||||||||||||||||
|
11. Determination of nicarbazin |
||||||||||||||||||||||||||||||||||||||||||||
|
12. Determination of vitamin A (retinol) |
||||||||||||||||||||||||||||||||||||||||||||
|
13. Determine of thiamine (vitamin B1, aneurine) |
||||||||||||||||||||||||||||||||||||||||||||
|
14. Determination of vitamin C |
||||||||||||||||||||||||||||||||||||||||||||
|
15. Determination of menadione (Vitamin K3) |
||||||||||||||||||||||||||||||||||||||||||||
|
16. Determination of hydrocyanic acid |
||||||||||||||||||||||||||||||||||||||||||||
|
17. Determination of free and total gossypol |
||||||||||||||||||||||||||||||||||||||||||||
|
18. Determination of theobromine |
||||||||||||||||||||||||||||||||||||||||||||
|
19. Determination of volatile mustard oil |
||||||||||||||||||||||||||||||||||||||||||||
|
20. Determination of buquinolate |
||||||||||||||||||||||||||||||||||||||||||||
|
21. Determination of sulphaquinoxaline |
||||||||||||||||||||||||||||||||||||||||||||
|
22. Determiation of furazolidone |
||||||||||||||||||||||||||||||||||||||||||||
|
23. Determination of aflatoxin B1 |
||||||||||||||||||||||||||||||||||||||||||||
|
24. Determination of urea |
||||||||||||||||||||||||||||||||||||||||||||
|
1. GENERAL PROVISIONS. |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Methods of analysis of the components of feeding stuffs are generally applicable to all feeding stuffs and premixtures. However, certain feeding stuffs, by reason of their inherent compositional characteristics, require certain modifications in analytical procedure. These have been provided for under the heading 'observations' in the description of methods. |
||||||||||||||||||||||||||||||||||||||||||||
|
When two or more methods may be used to determine a component of a feeding stuff, the choice of method shall, except where otherwise indicated, be left to the State Laboratory; the method used must however be indicated in the certificate of analysis. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Preparation of the sample for analysis |
||||||||||||||||||||||||||||||||||||||||||||
|
2.1 The sample shall be divided into two parts. One part shall be taken as it is for macroscopic and microscopic determinations and also for the determination of moisture. The other part shall be prepared as follows for chemical analysis. |
||||||||||||||||||||||||||||||||||||||||||||
|
2.2 Mix this part carefully on a clean dry surface. (If the sample is very moist it must be pre-dried to bring the moisture content down to a level between 8--12%. To do this, dry the sample at a suitable temperature for an adequate length of time). Divide it by means of a divider or by hand using the quartering method (which consists of taking portions in turn from two opposite sections). Reduce it by either of these methods to approximately 100 g and, if necessary, crush the reduced portion so that it will pass through a 1 mm round mesh sieve. Transfer this portion immediately to a dry container with an air-tight fitting. |
||||||||||||||||||||||||||||||||||||||||||||
|
2.3 The general provisions set out in 2.1 and 2.2 are applicable to all methods of analysis with the exception of those methods dealing with antibiotics, coccidiostats and vitamins. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
All reagents used should be of analytical quality. References to water mean distilled or de-ionised water. Where a solution of a reagent is mentioned without any other indication, this means a solution in distilled or de-ionised water. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Expression of Results |
||||||||||||||||||||||||||||||||||||||||||||
|
The result given in the certificate of analysis shall be the average value obtained on the basis of at least two determinations. Subject to special provisions, the result shall be expressed as a proportion by weight of the original sample as it was when it reached the State Laboratory. The result must not be given to more significant figures than the accuracy of the method of analysis allows. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. DETERMINATION OF VOLATILE NITROGENOUS BASES |
||||||||||||||||||||||||||||||||||||||||||||
|
(a) BY MICRODIFFUSION |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of volatile nitrogenous bases, expressed as ammonia, in feeding stuffs. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with water and the solution clarified and filtered. The volatile nitrogenous bases are displaced by microdiffusion using a solution of potassium carbonate, collected in a solution of boric acid and titrated with sulphuric acid. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Trichloroacetic acid solution, 20% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Indicator: dissolve 33 mg of bromocresol green and 65 mg of methyl red in 100 ml of 95--96% (v/v) ethanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Boric acid solution: in a 1 litre graduated flask dissolve 10 g of boric acid in 200 ml of 95--96% (v/v) ethanol and 700 ml of water. Add 10 ml of indicator (3 2). Mix and, if necessary, adjust the colour of the solution to light red by adding a solution of sodium hydroxide. 1 ml of this solution will fix a maximum of 300µg of NH3. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Saturated potassium carbonate solution: dissolve 100 g of potassium carbonate in 100 ml of boiling water. Leave to cool, filter. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sulphuric acid, 0.02 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Shaker: approximately 35--40 rpm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Glass or plastic Conway cells (see diagram). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Microburettes graduated in 1/100 ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, 10 g of sample and place with 100 ml of water in a 200 ml graduated flask. Shake in the shaker (4.1) for 30 minutes. Add 50 ml of trichloroacetic acid solution (3.1), make up to volume with water, shake vigorously and filter through a pleated filter. |
||||||||||||||||||||||||||||||||||||||||||||
|
Using a pipette, introduce 1 ml of boric acid solution (3.3) into the central part of the Conway cell (4.2) and 1 ml of the sample filtrate into the crown of the cell. Cover partially with the greased lid. Drop 1 ml of saturated potassium carbonate solution (3.4) quickly into the crown and close the lid so that the cell is airtight. Turn the cell carefully, rotating it in a horizontal plane so that the two reagents are mixed. Leave to incubate either for at least 4 hours at room temperature or for 1 hour at 40°C. Using a microburette (4.3), titrate the volatile bases in the boric acid solution with sulphuric acid 0.02 N (3.5). |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank test using the same procedure but without a sample to be analysed. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
1 ml of H2SO4 0.02 N corresponds to 0.34 mg of ammonia. |
||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between results of two parallel determinations carried out on the same sample should not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result for ammonia contents of less than 1.0%; |
||||||||||||||||||||||||||||||||||||||||||||
|
0.1%, in absolute value, for ammonia contents of 1.0% or more. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Observation |
||||||||||||||||||||||||||||||||||||||||||||
|
If the ammonia content of the sample exceeds 0.6%, dilute the initial filtrate. |
||||||||||||||||||||||||||||||||||||||||||||
|
(b) BY DISTILLATION |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of volatile nitrogenous bases, expressed as ammonia, in fish-meal containing practically no urea. It is applicable only to ammonia contents of less than 0.25%. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with water and the solution clarified and filtered. The volatile nitrogenous bases are displaced at boiling point by adding magnesium oxide and collected in a specific quantity of sulphuric acid, the excess of which is back-titrated with a solution of sodium hydroxide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Trichloroacetic acid solution, 20% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Magnesium oxide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Antifoam (eg silicone). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Sulphuric acid, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sodium hydroxide solution, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 0.3% (w/v) solution of methyl red in 95--96% (v/v) ethanol. |
||||||||||||||||||||||||||||||||||||||||||||
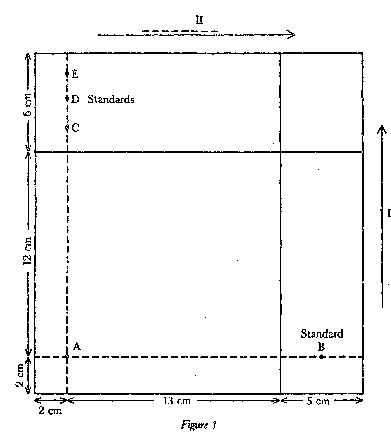
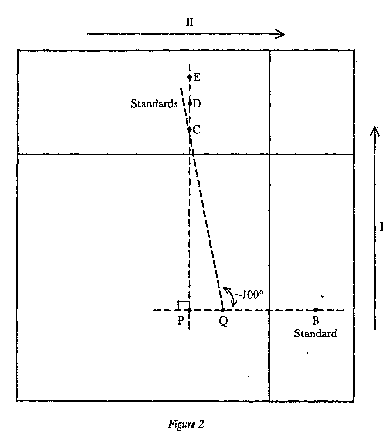
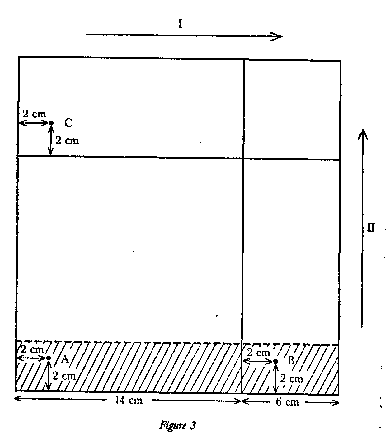
|
Conway Cell Scale 1/1 |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Shaker: approximately 35--40 rpm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Distilling apparatus of the Kjeldahl type. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 10 g of the sample, and place with 100 ml of water in a 200 ml graduated flask. Shake in the shaker (4.1) for 30 minutes. Add 50 ml of trichloroacetic acid solution (3.1), make up to volume with water, shake vigorously and filter through a pleated filter. |
||||||||||||||||||||||||||||||||||||||||||||
|
Take a quantity of clear filtrate appropriate for the presumed content of volatile nitrogenous bases (100 ml is usually suitable). Dilute to 200 ml and add 2 g of magnesium oxide (3.2) and a few drops of antifoam (3.3). The solution should be alkaline to litmus paper; otherwise add some magnesium oxide (3.2). Distil about 150 ml of the solution in the Kjeldahl apparatus (4.2) and collect the distillate in an Erlenmeyer flask containing an accurately measured volume (25--50 ml) of sulphuric acid 0.1 N (3.4). While distilling, avoid over-heating the sides of the flask. Boil the sulphuric acid solution for 2 minutes, cool it and back-titrate the excess sulphuric acid with the sodium hydroxide solution 0.1 N (3.5) in the presence of the methyl red indicator (3.6). |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank test using the same procedure but without a sample to be analysed. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
1 ml of H2SO4 0.1 N corresponds to 1.7 mg of ammonia. |
||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample should not exceed 10% relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. DETECTION AND IDENTIFICATION OF ANTIBIOTICS OF THE TETRACYCLINE GROUP |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To detect and identify antibiotics of the tetracycline group in feeding stuffs containing at least 0.1 mg/kg of antibiotics, in concentrates and in premixes. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with a mixture of methanol and hydrochloric acid. The extract and reference solutions for comparison are subjected to ascending paper chromatography. The antibiotics are detected and identified by comparing their Rf-values with those of the standard substances, either by fluorescence in UV light (high antibiotic contents) or by bioautography on an agar medium inoculated with B. cereus. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents and culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Eluent I: mixture of pure nitromethane/pure chloroform/1.3-dichloropropan-2-ol: 20/10/1.5 by volume, prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Eluent II: mixture of pure nitromethane/pure chloroform/2-picoline: 20/10/3 by volume. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Mixture of pure methanol/hydrochloric acid (d: 1.19): 98/2 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Hydrochloric acid, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Ammonia, d: 0.91. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Standard substances: chlortetracycline, oxytetracycline, tetracycline, the activity of which is expressed in terms of hydrochloride. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Micro-organism: B. cereus ATCC No 11.778 (NCIB 8849: NCTC 10320) |
||||||||||||||||||||||||||||||||||||||||||||
|
Maintenance of the parent strain, preparation of the spore suspension and inoculation of the culture medium: follow the directions given in 3.1 and 3.2 of the method for the determination of chlortetracycline, oxytetracycline and tetracycline contents by diffusion on agar; these instructions are the subject of Method 4 (a) paragraphs 3.1 and 3.2. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Adjust the pH to 5.8 immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Any commercial culture medium of similar composition and giving the same results may be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 0.1% (w/v) 2, 3, 5 triphenyltetrazolium chloride solution and 5% (w/v) glucose solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Apparatus for ascending paper chromatography (height of paper: 25 cm). |
||||||||||||||||||||||||||||||||||||||||||||
|
Schleicher and Schull paper (2040b or 2043b) or equivalent. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Centrifuge |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Incubator set at 30°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 U.V. lamp for the detection of fluorescence. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Glass plates approximately 20 x 30 cm for bioautography. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Stock solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
Using hydrochloric acid (3.6). prepare from the standard substances (3.8) solutions with concentrations corresponding to 500 µg per ml of chlortetracycline-HC1, of oxytetracycline-HC1, and of tetracycline-HC1. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Reference solutions for detections by UV light |
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the solutions (5.1) with the phosphate buffer solution (3.2) to obtain solutions with concentrations corresponding to 100 µg per ml of chlortetracycline-HC1, of oxytetracycline-HC1 and of tetracycline-HC1. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Reference solutions for detection by bioautograph |
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the solutions (5.1) with the phosphate buffer solution (3.2) to obtain solutions with concentrations corresponding to 5 µg per ml of chlortetracycline-HC1, of oxytetracycline-HC1 and of tetracycline-HC1. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
When the presumed antibiotic content is less than 10 mg/kg either the mixed sample or the finest fraction separated by sieving may be used, since the antibiotics are to be found mainly in this fraction. |
||||||||||||||||||||||||||||||||||||||||||||
|
Suspend the sample in the mixture (3.5) and centrifuge. Collect the supernatant liquid and use it directly or dilute, if necessary, with the mixture (3.5) to obtain antibiotic concentrations of approximately 100 µg per mg (6.1) and 5 µg per ml (6.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Detection and Mentification |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Immerse the paper in the buffer solution, pH 3.5 (3.1). Remove the excess liquid by pressing the paper between sheets of dry filter paper. Then place volumes of 0.01 ml of the reference solutions (5.2 and 5.3) and of the extract (6.1 and 6.2) on the paper. To give a good separation, the paper must have the correct moisture content; if necessary, leave to dry a little. |
||||||||||||||||||||||||||||||||||||||||||||
|
Develop by ascending chromatography. Use eluent 1 (3.3) for detection by bioautography and eluent II (3.4) for detection by UV light. When the solvent front has reached 15 to 20 cm (approx. 1 hour 30 minutes), stop chromatography and dry the paper. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Detection by UV light |
||||||||||||||||||||||||||||||||||||||||||||
|
If the antibiotic level is greater than 1 µg per cm2, after the chromatogram has been treated with ammonia vapours (3.7) golden yellow fluorescent spots will be seen on irradiation under the UV lamp (4.4). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 Detection by bioautography |
||||||||||||||||||||||||||||||||||||||||||||
|
Pour the culture medium (3.10); previously inoculated with B. cereus (3.9), into glass plates (4.5) and place the paper on the culture medium. After 5 minutes' contact, detach the paper and place it on another spot in the culture medium, where it will remain during the incubation period. |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubate overnight at 30°C. If an antibiotic of the tetracycline group is present, light inhibition zones will appear in the cloudy culture medium. |
||||||||||||||||||||||||||||||||||||||||||||
|
To fix the chromatogram, the solution (3.11) is vaporized on the paper, after incubation. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.4 Identification |
||||||||||||||||||||||||||||||||||||||||||||
|
The relative Rf values of antibiotics of the tetracycline group are given below. These values may vary slightly according to the quality of the paper and its moisture content: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
The antibiotic activity of the 'epi' compounds is less than that of the normal compounds. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. DETERMINATION OF CHLORTETRACYCLINE, OXYTETRACYCLINE AND TETRACYCLINE |
||||||||||||||||||||||||||||||||||||||||||||
|
(a) BY DIFFUSION ON AGAR |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the levels of chlortetracycline (CTC), oxytetracycline (OTC) and tetracycline (TC) in feedingstuffs. concentrates and premixes where more than 5 ppm are present. Contents of less than 5 mg/kg may be estimated by graphic interpolation. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
For contents of 50 ppm or less, the sample is extracted with dilute formamide. For contents greater than 50 ppm, it is extracted with a mixture of acetone, water and hydrochloric acid, for the determination of CTC, and with a mixture of methanol and hydrochloric acid for the determination of OTC and TC. |
||||||||||||||||||||||||||||||||||||||||||||
|
The extracts are then diluted and their antibiotic activity determined by measuring the diffusion of the CTC, OTC or TC on an agar medium seeded with B. cerus. The diffusion is made evident by the formation of inhibition zones in the presence of the micro-organism. The diameter of these zones is directly proportional to the logarithm of the antibiotic concentration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Micro-organism: B. cereus, ATCC No. 11.778 (NCIB 8849; NCTC 10320) |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Maintenance of the parent strain |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate with B. cereus a tube of sloped agar taken from culture medium (4.1) free from methylene blue and boric acid. Incubate overnight at approximately 30°C. Keep the culture in a refrigerator and re-inoculate sloped agar with it every 14 days. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Preparation of the spore suspension |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the bacteria from a tube of sloped agar (3.1) using 2--3 ml of physiological saline (4.5). With this suspension, seed a Roux flask containing 300 ml of culture medium (4.1), free from methylene blue and boric acid, with 3--4% agar concentration. Incubate for 3--5 days at 28--30°C, then collect the spores in 15 ml of ethanol (4.6) after checking sporulation under a microscope, and mix. This suspension will keep in a refrigerator for 5 months or more. |
||||||||||||||||||||||||||||||||||||||||||||
|
By preliminary tests on plates with the basic medium for the determination (4.1) establish the quantity of inoculum which, for the different concentrations of antiobiotic used, will give the largest possible inhibition zones that are still clear. This quantity is usually between 0.2--0.3 ml per 1000 ml. The culture medium is inoculated at between 50--60°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Culture media and reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Basic medium for the determination1 |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Any commercial culture medium of similar composition and giving the same results may be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Adjust to pH 5.8 before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Phosphate buffer solution, pH 5.5 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Phosphate buffer solution, pH 5.5, diluted to 1/10. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Phosphate buffer solution, pH 8 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Sterile physiological saline. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Ethanol solution, 20% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Hydrochloric acid, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Formamide solution, 70% (v/v): prepare fresh before use and adjust the pH to 4.5 using sulphuric acid approximately 2N. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.9 Mixture of pure acetone/water/hydrochloric acid (d: 1.19): 65/33/2 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.10 Mixture of pure methanol/hydrochloric acid (d:1.19): 98/2 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.11 Standard substances: CTC, OTC, TC, the activity of which is expressed in terms of hydrochloride. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Chlortetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Using hydrochloric acid (4.7), prepare from the standard solution (4.11) a stock solution with a concentration corresponding to 500 µg per ml of chlortetracycline-HC1. This solution will keep for one week in a refrigerator. From this stock solution, prepare a standard working solution S8 with a concentration corresponding to 0.2 µg per ml of chlortetracycline-HC1. Dilution is carried out using the phosphate buffer solution, pH 5.5, diluted to 1/10 (4.3), to which 0.01% of amido black has been added1. |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Amido black is used to make evident the inhibition zones of the standard solutions (blue rings). |
||||||||||||||||||||||||||||||||||||||||||||
|
Then prepare by successive dilutions (1 + 1), using the buffer solution (4.3), the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Oxytetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Proceeding as indicated in 5.1, prepare, from a stock solution with a concentration corresponding to 400 µg per ml of oxytetracycline-HC1, a standard working solution S8 containing 1.6 µg per ml of oxytetracycline-HC1, and the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Tetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Proceeding as indicated in 5.1, prepare, from a stock solution with a concentration corresponding to 500 µg per ml of tetracycline-HCl, a standard working solution S8 containing 1.0 µg per ml of tetracycline-HC1 and the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1 Contents of 50 mg/kg or less |
||||||||||||||||||||||||||||||||||||||||||||
|
To test sample add formamide (4.8) in the quantities indicated in the table below. Shake for 30 minutes on a shaking platform. Then dilute immediately with the phosphate buffer solution (4.3) according to the indications given in the table below to obtain the concentration U8. The formamide concentration of this solution must not exceed 40%. |
||||||||||||||||||||||||||||||||||||||||||||
|
Centrifuge or decant to obtain a clear solution. Then prepare the concentrations U4, U2 and U1 by successive dilutions (1 + 1) using the phosphate buffer solution (4.3). |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
(a) Take 20 ml of extract and make up to 100 ml in a graduated flask with the buffer solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
(b) Take 4 ml of extract and make up to 100 ml in a graduated flask with the buffer solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2 Contents greater than 50 mg/kg |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2.1 Chlortetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
To a test sample of 2--10 g, depending on the presumed antibiotic content of the sample or its manufacturer's guarantee, add 20 times its volume of mixture (4.9). Shake for 30 minutes on a shaking platform. The pH must remain below 3 during extraction; if necessary, readjust to pH 3 (using 10% acetic acid for mineral compounds). Take an aliquot part of the extract and adjust the pH to 5.5 using the phosphate buffer solution, pH 8 (4.4) in the presence of bromocresol green (turning from yellow to blue). Dilute, using the phosphate buffer solution, pH 5.5, diluted to 1/10 (4.3), to obtain the concentration U8 (see 6.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
Then prepare the concentrations U4, U2 and U1, by successive dilutions (1 + 1) using the phosphate buffer solution (4.3). |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2.2 Oxytetracycline and tetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Proceed as indicated in 6.2.1, using the mixture (4.10) instead of the mixture (4.9). |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination method |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Inoculation of the culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate at 50--60°C the basic medium for the determination (4.1) with the spore suspension (3.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Preparation of the trays |
||||||||||||||||||||||||||||||||||||||||||||
|
Diffusion on agar is carried out in trays using 4 concentrations of the standard solution (S8, S4, S2, S1) and 4 concentrations of the extract (U8, U4, U2, U1). The 4 concentrations of extract and of standard solution must be placed in each tray. |
||||||||||||||||||||||||||||||||||||||||||||
|
Choose trays, therefore, which are large enough to allow at least 8 holes 10--13 mm in diameter to be made in the agar medium. Calculate the quantity of inoculated culture medium (7.1) needed to provide a uniform covering approximately 2 mm thick. The test should preferably be carried out on trays consisting of flat glass plates fitted with a perfectly level aluminium or plastic ring, 200 mm in diameter and 20 mm high. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into the holes accurately measured quantities of between 0.10--0.15 ml of antibiotic solution, depending on the diameter of the holes. |
||||||||||||||||||||||||||||||||||||||||||||
|
For each sample, repeat the diffusion at least 4 times with each concentration so that each determination comprises an evaluation of 32 inhibition zones. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 Incubation |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubate the trays for approximately 18 hours at 28--30°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Evaluation |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the diameter of the inhibition zones, preferably by projection. Record the measurements on semi-logarithmic paper, plotting the logarithm of the concentrations against the diameter of the inhibition zones. Trace the lines of the standard solution and of the extract. Provided there is no interference, the two lines will be parallel. |
||||||||||||||||||||||||||||||||||||||||||||
|
The logarithm of the relative activity is calculated by using the following formula:-- |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
Real activity = presumed activity x relative activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10%, relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
(b) BY TURBIDIMETRY |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the levels of chlortetracycline (CTC), oxytetracycline (OTC) and tetracycline (TC) where concentrations are greater than 1 g per kg, provided there is no interference from other substances clouding the extracts. This method is quicker than diffusion on agar. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
For the determination of CTC, the sample is extracted with a mixture of acetone, water and hydrochloric acid, and with a mixture of methanol and hydrochloric acid for the determination of OTC and TC. |
||||||||||||||||||||||||||||||||||||||||||||
|
The extracts are then diluted and their antibiotic effect determined by measuring the light transmission of a culture medium which has been seeded with Staphylococcus aureus and to which the antibiotic has been added. The light transmission depends on the antibiotic concentration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Micro-organism: Staphylococcus aureus K 1411 (NCIB 11182; NCTC 10988) |
||||||||||||||||||||||||||||||||||||||||||||
|
1 This strain, isolated by the LUFA at Kiel, grows more rapidly than S. aureus ATCC 6538P. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Maintenance of the parent strain |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate with S. aureus a tube of sloped agar taken from the culture medium (4.1), to which 1.5--3% of agar has been added (depending on the quality). Incubate overnight at 37°C. Keep the culture in a refrigerator and re-inoculate sloped agar with it every 4 weeks. At the same time prepare sub-cultures for laboratory use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Preparation of the inoculum |
||||||||||||||||||||||||||||||||||||||||||||
|
24 hours before use, re-inoculate sloped agar with a sub-culture and incubate overnight at 37°C. Suspend all the culture contained in a tube of agar in approximately 2 ml of the basic medium (4.1), then transfer the suspension under sterile conditions into approximately 100 ml of the same basic medium (4.1). Incubate in a water bath at 37°C until the growth of the strain enters its logarithmic phase (1 hour 30 minutes to 2 hours). |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Culture media and reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.2. Phosphate buffer solution, pH 4.5 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.3. Hydrochloric acid, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4. Mixture of pure acetone/water/hydrochloric acid (d: 1.19): 65/33/2 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5. Mixture of pure methanol/hydrochloric acid (d: 1.19): 98/2 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6. Formaldehyde solution, approximately 10% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7. Standard substances: CTC, OTC, TC, the activity of which is expressed in terms of hydrochloride. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solution |
||||||||||||||||||||||||||||||||||||||||||||
|
Using hydrochloric acid (4.3), prepare from the standard substance (4.7) a stock solution with a concentration corresponding to 400--500 µg per ml of CTC-HC1, OTC-HC1 or TC-HC1. This solution will keep for one week in a refrigerator. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1. Chlortetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Place a 1--2 g test sample in a 200 or 250 ml graduated flask. Add approximately 100 ml of the mixture (4.4) and shake for 30 minutes on a shaking platform. Make up to volume with the phosphate buffer solution, pH 4.5 (4.2). Mix and leave to settle. |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2. Oxytetracycline and tetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Place a 1--2 g test sample in a 200 or 250 ml graduated flask. Add approximately 100 ml of the mixture (4.5) and shake for 30 minutes on a shaking platform. Make up to volume with the phosphate buffer solution, pH 4.5 (4.2). Mix and leave to settle. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination method |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1. Preparation of the standard series and of the extract |
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the standard solution (5) and the extract (6) with the phosphate buffer solution, pH 4.5 (4.2), to obtain a series of concentrations. For each determination, a calibration curve is drawn from the respective concentration, permitting the interpolation of at least two values relating to the extract. The dilutions should be chosen according to the conditions under which the strain is grown, which may vary from one laboratory to another. The procedure is generally the following: |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1.1. Chlortetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the standard solution (5) with the phosphate buffer solution (4.2) to obtain a standard working solution with a concentration corresponding to 0.2 µg per ml of CTC-HC1. Then, using the phosphate buffer solution (4.2), prepare in test tubes, as indicated below, 6 dilutions, each dilution in duplicate. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the extract (6.1) with the phosphate buffer solution (4.2) to obtain a presumed CTC-HC1 concentration of 0.12 µg per ml. Place 1 ml of this solution in each of 2 tubes; and 0.75 ml (=0.09 µg) in each of 2 other tubes. Make the volume of the latter 2 tubes up to 1 ml with the phosphate buffer solution (4.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1.2 Oxytetracycline and tetracycline |
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the standard solution (5) with the phosphate buffer solution (4.2) to obtain a standard working solution with a concentration corresponding to 0.6 µg per ml of OTC-HC1 or of TC-HC1. Then, using the phosphate buffer solution (4.2), prepare in test tubes, as indicated below, 7 dilutions, each dilution in duplicate. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute the extract (6.2) with the phosphate buffer solution (4.2) to obtain a presumed OTC-HC1 or TC-HC1 concentration of 0.48 µg per ml. Place 1 ml of this solution in each of 2 tubes, and 0.5 ml (=0.24 µg) in each of 2 other tubes. Make the volume of the latter 2 tubes up to 1 ml with the phosphate buffer solution (4.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2. Inoculation of the culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate the basic medium for the determination (4.1) with the inoculum (3.2) to obtain with the photometer at 590 nm 85% light transmission in a 5 cm cell or 92% transmission in a 2 cm cell, the apparatus being set at 100% transmission on the non-inoculated basic medium (4.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3. Seeding |
||||||||||||||||||||||||||||||||||||||||||||
|
Place 9 ml of the inoculated culture medium (7.2) in each tube (7.1.1 or 7.1.2). The tubes must be filled under clean but not necessarily sterile conditions. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.4. Incubation |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubation must be carried out in a water bath whose temperature is kept uniform at 37°C±0.1°C by stirring. The incubation period chosen (generally 2 hours 30 minutes to 3 hours) must be such that it will be possible to trace transmission curves with gradients suitable for accurate measurement. Then block further growth by rapidly injecting 1 ml of formaldehyde solution (4.6) into each tube. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.5. Measurement of growth |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the transmissions with the photometer at 590 nm, setting the apparatus at 100% transmission on the clearest standard solution (corresponding to the highest antibiotic content). Since the different tubes will show slight differences of turbidity, at least 2 cm, and preferably 5 cm, cells should be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Trace the calibration curve on millimetre graph paper by plotting the photometric transmissions against the antibiotic concentrations. Interpolate on the curve the transmission values of the extract. Calculate the antibiotic content of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10%, relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. DETERMINATION OF OLEANDOMYCIN |
||||||||||||||||||||||||||||||||||||||||||||
|
by diffusion on agar |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine, even in the presence of tetracyclines, the oleandomycin content of feedingstuffs, concentrates and premixes, where more than 0.5 ppm mg/kg are present. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with a dilute methanol solution of tri (hydroxymethylamino) methane. After centrifuging, the extract is diluted and its antibiotic activity determined by measuring the diffusion of the oleandomycin on an agar medium seeded with B. cereus. The diffusion is made evident by the formation of inhibition zones in the presence of the micro-organism. The diameter of these zones is directly proportional to the logarithm of the antibiotic concentration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Micro-organism: B. cereus K 250 TR1 (resistant to tetracyclines) (NCIB 11183; NCTC 10989) |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Strain isolated by the LUFA at Kiel. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Maintenance of the parent strain |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate with B. cereus a tube of sloped agar taken from the culture medium (4.1) to which 100 µg per 5 ml oxytetracycline has been added. Incubate overnight at approximately 30°C. Keep the culture in a refrigerator and re-inoculate sloped agar with it every 4 weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Preparation of the spore suspension |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the bacteria from a tube of sloped agar (3.1) using approximately 3 ml of physiological saline (4.3). With this suspension, seed a Roux flask containing 300 ml of culture medium (4.1) which has a 3--4% agar concentration. Incubate for 3--5 days at 28--30°C, then collect the spores in 15 ml of ethanol (4.4) after checking sporulation under a microscope, and mix. This suspension will keep in a refrigerator for 5 months or more. |
||||||||||||||||||||||||||||||||||||||||||||
|
By preliminary tests on plates with the basic medium for the determination (4.2), establish the quantity of inoculum which, for the different concentrations of oleandomycin used will give the largest possible inhibition zones that are still clear. This quantity is usually between 0.1--0.2 ml per 1000 ml. The culture medium is inoculated at 60°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Culture media and reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Medium for maintenance of the parent strain1 |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Any commercial culture medium of similar composition and giving the same results may be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Basic medium for the determination1 |
||||||||||||||||||||||||||||||||||||||||||||
|
Medium (4.1) adjusted to pH 8.8. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Sterile physiological saline. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Ethanol solution, 20% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Tri (hydroxymethylamino) methane solution, 0.5% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Extraction solution |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Standard substance: oleandomycin of known activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solution |
||||||||||||||||||||||||||||||||||||||||||||
|
Dissolve some of the standard substance (4.8) in 5 ml of methanol (4.5) and dilute with the solution (4.6) to obtain an oleandomycin concentration of 100 µg per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
From this stock solution, prepare a standard working solution S8 containing 0.1 µg per ml of oleandomycin by diluting with the solution (4.6). Then prepare by successive dilutions (1 + 1), using the solution (4.6), the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Take a test sample of 2--10 g, depending on the presumed oleandomycin content of the sample, add 100 ml of the solution (4.7) and shake for 30 minutes on a shaking platform. |
||||||||||||||||||||||||||||||||||||||||||||
|
Centrifuge, take an aliquot part of the extract and dilute with the solution (4.6) to obtain a presumed oleandomycin concentration of 0.1 µg per ml (= U8). Then prepare the concentrations U4, U2 and U1 by successive dilutions (1 + 1) using the solution (4.6). |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination method |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Inoculation of the culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate at 60°C the basic medium for the determination (4.2) with the spore suspension (3.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Preparation of the trays |
||||||||||||||||||||||||||||||||||||||||||||
|
Diffusion on agar is carried out in trays using 4 concentrations of the standard solution (S8, S4, S2, S1 ) and four concentrations of the extract (U8, U4, U2, U1,). The 4 concentrations of standard solution and of extract must be placed in each tray. |
||||||||||||||||||||||||||||||||||||||||||||
|
Choose trays, therefore, which are large enough to allow at least 8 holes 10--13 mm in diameter to be made in the agar medium. Calculate the quantity of inoculated culture medium (7.1) needed to provide a uniform covering approximately 2 mm thick. The test should preferably be carried out on trays consisting of flat glass plates fitted with a perfectly level aluminium or plastic ring 200 mm in diameter and 200 mm high. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into the holes accurately measured quantities of between 0.10--0.15 ml of antibiotic solution, depending on the diameter of the holes. |
||||||||||||||||||||||||||||||||||||||||||||
|
For each sample repeat the diffusion at least 4 times with each concentration so that each determination comprises an evaluation of 32 inhibition zones. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 Incubation |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubate the trays for approximately 18 hours at 28--30°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Evaluation |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the diameter of the inhibition zones, preferably by projection. Record the measurements on semi-logarithmic paper, plotting the logarithm of the concentrations against the diameter of the inhibition zones. Trace the lines of the standard solution and of the extract. Provided there is no interference, the two lines will be parallel. |
||||||||||||||||||||||||||||||||||||||||||||
|
The logarithm of the relative activity is calculated by using the following formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
Real activity = presumed activity x relative activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parrallel determinations carried out on the same sample must not exceed 10% relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. DETERMINATION OF TYLOSIN |
||||||||||||||||||||||||||||||||||||||||||||
|
by diffusion on agar |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the tylosin content of feedingstuffs, concentrates and premixes where more than 2 mg/kg are present. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is treated with a pH 8 phosphate buffer solution, previously heated to 80°C, and then extracted with methanol. After centrifuging, the extract is diluted and its antibiotic activity determined by measuring the diffusion of the tylosin on an agar medium seeded with Sarcina lutea. The diffusion is made evident by the formation of inhibition zones in the presence of the micro-organism. The diameter of these zones is directly proportional to the logarithm of the antibiotic concentration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Micro-organism: Sarcina lutea ATCC No. 9341 (NCIB 8553; NCTC 8340) |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Maintenance of the parent strain |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate with Sarcina lutea a tube of sloped agar taken from the culture medium (4.1), adjust to pH 7.0. Incubate overnight at approximately 35°C. Keep the culture in a refrigerator and reinoculate sloped agar with it every month. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Preparation of the bacteria suspension |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the bacteria from a recently prepared tube of sloped agar (3.1) using 2--3 ml of physiological saline (4.4). With this suspension seed a Roux flask containing 250 ml of the culture medium (4.1), adjusted to pH 7.0. Incubate for 24 hours at 35°C, then collect the bacteria in 25 ml of physiological saline (4.4). Mix, and dilute this suspension to obtain approximately 75% light transmission at 650 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
If kept in a refrigerator this suspension may be used for one week. |
||||||||||||||||||||||||||||||||||||||||||||
|
By preliminary tests on plates with the basic medium for the determination (4.1), establish the quantity of inoculum which, for the different concentrations of tylosin used, will give the largest possible inhibition zones that are still clear. The culture medium is inoculated at 48--50°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Culture media and reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Basic medium for the determination1 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Adjust immediately before use to pH 7.0 for maintenance of the parent strain and preparation of the bacteria suspension, and to pH 8.0 for the determination. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Phosphate buffer solution, pH 8 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Phosphate buffer solution, pH 7 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Sterile physiological saline. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Methanol solution, 10% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Mixture of phosphate buffer solution (4.2)/pure methanol: 60/40 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Standard substance: tylosin of known activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
Dry the standard substance (4.8) for 3 hours at 60°C in a vacuum oven (5 mm of mercury). Weigh 10--50 mg into a graduated flask, dissolve in 5 ml of methanol (4.5) and dilute the solution with the phosphate buffer solution, pH 7 (4.3), to obtain a tylosin-base concentration of 1000 µg per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare a standard working solution S8 containing 2 µg per ml of tylosin base from this stock solution by diluting with the mixture (4.7). |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Any commercial culture medium of similar composition and giving the same results may be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
Then prepare by successive dilutions (1 + 1), using the mixture (4.7), the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
For concentrates, take a 10 g test sample; for premixes and feedingstuffs, a 20 g test sample. Add 60 ml of phosphate buffer solution, pH8 (4.2), previously heated to 80°C, and mix for 2 minutes (domestic mixer, Ultra-turrax, etc.). Leave to stand for 10 minutes, add 40 ml of methanol (4.5) and mix for 5 minutes. Centrifuge the extract and dilute an aliquot part with the mixture (4.7) to obtain a presumed tylosin concentration of 2 µg per ml (=U8). Then prepare the concentrations U4, U2 and U1, by successive dilutions (1 + 1) using the mixture (4.7). |
||||||||||||||||||||||||||||||||||||||||||||
|
For contents of less than 10 ppm, evaporate the extract until dry in a rotary evaporator at 35°C and dissolve the residue in 10% methanol (4.6). |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination method |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Inoculation of the culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate at 48--50°C the basic medium for the determination (4.1), adjust to pH 8.0, with the bacteria suspension (3.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Preparation of the trays |
||||||||||||||||||||||||||||||||||||||||||||
|
Diffusion on agar is carried out in trays using 4 concentrations of the standard solution (S8, S4, S2, S1) and 4 concentrations of the extract (U8, U4, U2, U1). The 4 concentrations of standard solution and of extract must be placed in each tray. |
||||||||||||||||||||||||||||||||||||||||||||
|
Choose trays, therefore, which are large enough to allow at least 8 holes 10--13 mm in diameter to be made in the agar medium. Calculate the quantity of inoculated culture medium (7.1) needed to provide a uniform covering approximately 2 mm thick. The test should preferably be carried out on flat trays consisting of glass plates fitted with a perfectly level aluminium or plastic ring, 200 mm in diameter and 20 mm high. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into the holes acurately measured quantities of between 0.10--0.15 ml of antibiotic solution, depending on the diameter of the holes. |
||||||||||||||||||||||||||||||||||||||||||||
|
For each sample repeat the diffusion at least 4 times with each concentration so that each determination comprises an evaluation of 32 inhibition zones. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 Incubation |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubate the trays overnight at 35--37°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Evaluation |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the diameter of the inhibition zones, preferably by projection. Record the measurements on semi-logarithmic paper, plotting the logarithm of the concentrations against the diameter of the inhibition zones. Trace the lines of the standard solution and of the extract. Provided there is no interference the two lines will be parallel. |
||||||||||||||||||||||||||||||||||||||||||||
|
The logarithm of the relative activity is calculated by using the following formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
Real activity = presumed activity x relative activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10% relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. DETERMINATION OF VIRGINIAMYCIN |
||||||||||||||||||||||||||||||||||||||||||||
|
by diffusion on agar. |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the virginiamycin content of feeding stuffs, concentrates and premixes where more than 2 mg/kg are present. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with a "Tween 80" methanol solution. After centrifuging or filtering, the extract is diluted and its antibiotic activity determined by measuring the diffusion of the virginiamycin on an agar medium seeded with Sarcina lutea. The diffusion is made evident by the formation of inhibition zones in the presence of the micro-organism. The diameter of these zones is directly proportional to the logarithm of the antibiotic concentration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Micro-organism: Sarcina lutea ATCC No 9341 (NCIB 8553, NCTC 8340) |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Maintenance of the parent strain |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate with S. Lutea a tube of sloped agar taken from the culture medium (4.1). Incubate overnight at approximately 35°C. Keep the culture in a refrigerator and re-inoculate sloped agar with it every 14 days. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Preparation of the bacteria suspension |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the bacteria from a recently prepared tube of sloped agar (3.1) using 2--3 ml of physiological saline (4.3). With this suspension, seed a Roux flask containing 250 ml of the culture medium (4.1). Incubate for 24 hours at 35°C. then collect the bacteria in 25 ml of physiological saline (4.3). Mix, and dilute this suspension to obtain approximately 75% light transmission at 650 nm. If kept in a refrigerator this suspension may be used for 1 week. |
||||||||||||||||||||||||||||||||||||||||||||
|
By preliminary tests on plates with the basic medium for the determination (4.1). establish the quantity of inoculum which, for the different concentrations of virginiamycin used, will give the largest possible inhibition zones that are still clear. The culture medium is inoculated at 48-50°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Culture media and reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Basic medium for the determination1 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Adjust the pH to 6.5 before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Phosphate buffer solution, pH 6 |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Sterile physiological saline |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Methanol |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Mixture of phosphate buffer solution (4.2)/pure methanol: 80/20 by volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 "Tween 80" methanol solution, 0.5% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Standard substance: virginiamycin of known activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
1 Any commercial culture medium of similar composition and giving the same results may be used. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Standard solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare a methanol solution of the standard substance (4.7) containing 800 µg per ml of virginiamycin. From this stock solution prepare a standard working solution S8 containing 1µg per ml of virginiamycin by diluting with the mixture (4.5). Then prepare by successive dilutions (1 + 1), using the mixture (4.5), the following concentrations: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
6. Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1 Products with a virginiamycin content of 50 mg/kg or less |
||||||||||||||||||||||||||||||||||||||||||||
|
Take a test sample of 10--20 g, add 100 ml of the solution (4.6) and shake for 30 minutes on a shaking platform. Centrifuge or filter, take 20 ml of the clear solution and evaporate until dry in a rotary evaporator. Dissolve the residue in 20 ml or more of the mixture (4.5) to obtain a presumed virginiamycin concentration of 1 µg per ml (= U8). Then prepare the concentrations U4, U2 and U1 by successive dilutions (1 + 1) using the mixture (4.5). |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2 Products with a virginiamycin content greater than 50 mg/kg |
||||||||||||||||||||||||||||||||||||||||||||
|
Take a test sample of 1--10 g, add 100 ml of solution (4.6) and shake for 30 minutes on a shaking platform. Centrifuge or filter, then dilute with the mixture (4.5) to obtain a presumed virginiamycin concentration of 1 µg per ml (= U8). Then prepare the concentrations U4 U2 and U1 as indicated in 6.1. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Determination method |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Inoculation of the culture medium |
||||||||||||||||||||||||||||||||||||||||||||
|
Inoculate the basic medium for the determination (4.1) at 48--50°C with the suspension of bacteria (3.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Preparation of the trays |
||||||||||||||||||||||||||||||||||||||||||||
|
Diffusion on agar is carried out in trays using 4 concentrations of the standard solution (S8, S4, S2, S1) and 4 concentrations of the extract (U8, U4, U2, U1). The 4 concentrations of the standard solution and of extract must be placed in each tray. |
||||||||||||||||||||||||||||||||||||||||||||
|
Choose trays, therefore, which are large enough to allow at least 8 holes 10--13 mm in diameter to be made in the agar medium. Calculate the quantity of inoculated culture medium (7.1) needed to provide a uniform covering approximately 2 mm thick. The test should preferably be carried out on flat trays consisting of glass plates fitted with a perfectly level aluminium or plastic ring, 200 mm in diameter and 20 mm high. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into the holes accurately measured quantities of between 0.10--0.15 ml of antibiotic solution, depending on the diameter of the holes. |
||||||||||||||||||||||||||||||||||||||||||||
|
For each sample, repeat the diffusion at least 4 times with each concentration so that each determination comprises an evaluation of 32 inhibition zones. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 Incubation |
||||||||||||||||||||||||||||||||||||||||||||
|
Incubate the trays for approximately 18 hours at 28--30°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
8 Evaluation |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the diameter of the inhibition zones, preferably by projection. Record the measurements on semi-logarithmic paper, plotting the logarithm of the concentrations against the diameter of the inhibition zones. Trace the lines of the standard solution and of the extract. Provided there is no interference, the two lines will be parallel. |
||||||||||||||||||||||||||||||||||||||||||||
|
The logarithm of the relative activity is calculated by using the following formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
Real activity = presumed activity x relative activity. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10% relative to the higher result. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. DETERMINATION OF AMPROLIUM |
||||||||||||||||||||||||||||||||||||||||||||
|
(Chloride hydrochloride of 1-(4-amino-2-propyl-5-pyrimidylmethyl)-2-picolinium) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of amprolium in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 40 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with diluted methanol. The extract is purified on a column of aluminium oxide and treated with a methanol solution of 2, 7-dihydroxyn aphthalene, potassium ferricyanide, potassium cyanide and sodium hydroxide, forming a purple colour complex. Amprolium is determined by spectrophotometry at 530 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Diluted methanol: mix two volumes of methanol (3.1) with one volume of water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Potassium ferricyanide (K3Fe (CN)6) solution, 0.2% (w/v). This solution is stable for 2 weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Potassium cyanide solution, 1% (w/v). This solution is stable for two weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sodium hydroxide solution, 1.125% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Methanolic sodium hydroxide solution: dilute 15 ml of the solution (3.5) to 200 ml with methanol (3.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 2, 7--dihydroxynaphthalene solution, 0.0025% (w/v): dissolve 25 mg of 2, 7-dihydroxynaphthalene in methanol (3.1) and make it up to 1000 ml with methanol (3.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Colour reagent: transfer 90 ml of 2, 7-dihydroxynaphthalene solution (3.7) to a conical flask (4.1), add 5 ml of potassium ferricyanide solution (3.3) and mix well. Then add 5 ml of potassium cyanide solution (3.4), stopper the flask and mix well. Leave to stand for 30--35 minutes, add 100 ml of methanolic sodium hydroxide solution (3.6), mix and filter through a filter crucible (4.3). Use this reagent in the 75 minutes following filtration. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Aluminium oxide for column chromatography: before use, stir 100 g of aluminium oxide with 500 ml of water for 30 minutes, filter the slurry, wash the aluminium oxide on the filter 3 times with 50 ml of methanol (3.1), drying each time by suction, leave to stand overnight and then dry for 2 hours at 100°C in a vacuum drier. Put in a desiccator to cool. Check strength by subjecting a specified quantity of standard solution (3.11) to analysis, starting from point 5.2. The recovery rate of the amprolium must be 100%±4%. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Standard substance: pure amprolium complying with the characteristics below: |
||||||||||||||||||||||||||||||||||||||||||||
|
Melting point (decomposition): 248°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
Molecular extinction co-efficient at both 265 and 235 nm in water: 11.0 x 103. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Standard solution: weigh out to within 0.1 mg, 50 mg of standard substance (3.10). Dissolve in diluted methanol (3.2) in a 500 ml volumetric flask, make up the volume with the same solvent and mix. Dilute 10.0 ml to 50 ml with diluted methanol (3.2) in a volumetric flask and mix well. 1 ml of this solution contains 20 µg of amprolium. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flasks with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Stirrer. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Filtering crucible, porosity G3, diameter: 60 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Glass tube for chromatography (interior diameter: approximately 9 mm, length: 400--500 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Centrifuge. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Spectrophotometer with suitable cells (10 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Extraction and purification |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1.1 Feeding stuffs and premixtures |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 10 g of the finely divided and mixed sample. For premixtures weigh 3--6 g, to the nearest mg. Place the test portion in a conical flask (4.1) and add exactly 100 ml of diluted methanol (3.2). Shake for 60 minutes and filter. Dilute with diluted methanol (3.2) if necessary to obtain a solution containing 5--15 µg of amprolium per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
Insert a cotton wool plug into the lower end of a chromatographic tube (4.4), and tamp in 5 g of alumium oxide (3.9) and then run in 25.0 ml of the extract. Let the liquid run through the column, discard the first 5 ml and collect the next 12 ml in a graduated test tube. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1.2 Concentrates |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 0.5 g of the finely divided and mixed sample, place it in a conical flask (4.1), add 250 ml of diluted methanol (3.2) shake or stir for 60 minutes and filter. Dilute 5.0 ml of the Filtrate to 200 ml with diluted methanol (3.2) in a volumetric flask. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Development of colour and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer 5.0 ml of the solution obtained in 5.1.1 or 5.1.2 into centrifuge tube A (4.5). Place 5.0 ml of diluted methanol (3.2) in centrifuge tube B (4.5). Add to each tube 10.0 ml of colouring reagent (3.8), stopper the tubes, mix and leave to stand for 18 minutes. Then centrifuge for 3 minutes in order to obtain a clear solution and decant solutions A and B in conical flasks (4.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
Immediately measure the optical density of solution A at 530 nm in the spectrophotometer (4.6) using solution B as a control. Determine the quantity of amprolium by referring to the calibration curve (5.3). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into centrifuge tubes (4.5) volumes of 1.0, 2.0, 3.0, 4.0 and 5.0 ml respectively of the standard solution (3.11). Make the volumes of the first four tubes up to 5.0 ml with diluted methanol (3.2). Add to all five tubes 10.0 ml of coloring reagent (3.8), stopper the tubes, mix and leave to stand for 18 minutes. Then centrifuge for 3 minutes and decant the solutions into conical flasks (4.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
Immediately measure the optical density of the solutions at 530 nm in the spectrophotometer (4.6), using a mixture of 5 ml diluted methanol (3.2) and 10 ml of colour reagent (3.8) as a control. Plot the calibration curve, using the optical density values as the ordinates and the corresponding quantities of amprolium in mg as the abscissae. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1 Feeding stuffs and premixtures |
||||||||||||||||||||||||||||||||||||||||||||
|
The amprolium content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity of amprolium in mg as determined by photometric measurement; |
||||||||||||||||||||||||||||||||||||||||||||
|
W = weight of the test portion in grams; |
||||||||||||||||||||||||||||||||||||||||||||
|
F = coefficient of dilution (possibly worked out in 5.1.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2 Concentrates |
||||||||||||||||||||||||||||||||||||||||||||
|
The amprolium content per cent of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity of amprolium in mg as determined by photometric measurement; |
||||||||||||||||||||||||||||||||||||||||||||
|
W = weight of the test portion in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value, for amprolium contents below 100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100 and 5000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value, for contents between 5000 and 10000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
9. DETERMINATION OF ETHOPABATE |
||||||||||||||||||||||||||||||||||||||||||||
|
(methyl-4-acetamido-2-ethoxybenzoate) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of ethopabate in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 2 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with diluted methanol. The solution is acidified and extracted with chloroform. The chloroform extract is washed first with an alkaline solution and then with water. The purified extract is concentrated, the ethopabate is hydrolysed with diluted hydrochloric acid. The animo derivative thus formed is diazotised and coupled with 2-aminoethyl-1-naphthylamine dihydrochloride. The colored complex is extracted with butanol and the optical density of the solution is measured at 555 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Methanol solution, 50% (v/v): mix equal volumes of methanol (3.1) and water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Hydrochloric acid, d: 1.18. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Hydrochloric acid solution, 10% (v/v): dilute 10.0 ml of hydrochloric acid (3.3) to 100 ml with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Hydrochloric acid, approximately 0.3 N: dilute 25 ml of hydrochloric acid (3.3) to 1,000 ml with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Chloroform. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Sodium carbonate solution, 4% (w/v): dissolve 40.0 g of anhydrous sodium carbonate in water and make up to 1,000 ml with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Sodium nitrate solution 0.2% (w/v): dissolve 100 mg of sodium nitrate in water and make up to 50 ml with water in a volumetric flask. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Ammonium sulphamate solution 1.0% (w/v): dissolve 500 mg of ammonium sulphamate in water and make up to 50 ml with water in a volumetric flask. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 2-aminoethyl-1-naphthylamine dihydrochloride solution, 0.2% (w/v): dissolve 100 mg of 2-aminoethyl-1-naphthylamine dihydrochloride in water and make up to 50 ml with water in a volumetric flask. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Anhydrous sodium chloride. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.12 n-butanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Standard substance: ethopabate. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Standard solutions: |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14.1 Solution of 0.040 mg of ethopabate per ml: weigh out 40 mg to within 0.1 mg of standard substance (3.13). Dissolve in methanol (3.2) in a 100 ml volumetric flask; make up the volume with the same solvent and mix. Dilute 10.0--100 ml with methanol (3.2) in a volumetric flask and mix. This solution is stable for a month. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14.2 Solution of 0.016 mg of ethopabate per 20 ml: dilute 5.0 ml of the solution (3.14.1) to 250 ml with methanol (3.2) in a volumetric flask and mix well. Prepare before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flasks, with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Separating funnels, with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Shaker. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Rotary vacuum evaporator. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Water bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Centrifuge. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Spectrophotometer with suitable cells (10 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, a quantity of the finely divided and mixed sample, containing about 80 µg of ethopabate. Place the test portion in a conical flask (4.1) and add 100.0 ml of diluted methanol (3.2). Mix, stopper the flask and shake for 1 hour with the aid of a shaker (4.3). Decant, filter and discard the first few ml of the filtrate. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Purification |
||||||||||||||||||||||||||||||||||||||||||||
|
NB All operations under this point must be carried out rapidly. Transfer 20.0 ml of the clear extract into a separating funnel (4.2), add 5.0 ml of hydrochloric acid (3.4) and 20.0 ml of chloroform (3.6). Shake, first carefully and then vigorously for 3 minutes. Leave to stand until the zones separate and collect the chloroform phase in a second separating funnel (4.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
Extract the acid phase twice more with 20.0 ml of chloroform (3.6). Collect the chloroform extracts in the second separating funnel and discard the acid phase. Add to the combined chloroform solution 10 ml of sodium carbonate solution (3.7), shake for 3 minutes and leave to stand until the phases separate. Collect the chloroform phase in a third separating funnel (4.2) and discard the aqueous phase. Add to the chloroform solution 10 ml of sodium carbonate solution (3.7), shake for 3 minutes and leave to stand until the phases separate. |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the chloroform phase in a fourth separating funnel (4.2) wash twice consecutively with 25.0 ml of water each time, separate the aqueous phases and quantitatively collect the chloroform extract in a round bottomed flask. Combine the aqueous phases together, in one of the separating funnels; rinse each empty funnel with a few ml chloroform; shake the aqueous phase with the same few ml chloroform, allow phases to separate, and transfer the chloroform phase to the chloroform extract collected in the flask. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Hydrolysis |
||||||||||||||||||||||||||||||||||||||||||||
|
Evaporate the chloroform extract down to about 2 ml on a 50°C water bath with the aid of the rotary vacuum evaporator (4.4). Dissolve the residue in 2-3 ml of methanol (3.1), and transfer quantitatively the solution in a centrifuge tube (4.6) with the aid of two 10 ml portions and one 5 ml portion of 0.3 N hydrochloric acid (3.5). Add a few glass beads, shake well, plunge the tube in a bath of boiling water and keep it there for 45 minutes. Then cool under a stream of cold running water. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Development of colour and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 1.0 ml of sodium nitrate solution (3.8), stir and leave to stand for 2 minutes. Add 1.0 ml of ammonium sulphamate solution (3.9), shake and leave to stand for 2 minutes. Add 1.0 ml of 2-aminoethyl-1-naphthylamine dihydrochloride solution (3.10), stir and leave to stand for 10 minutes. Add 5.0 g of sodium chloride (3.11) and 10.0 ml of n-butanol (3.12), shake vigorously until the sodium chloride has completely dissolved. |
||||||||||||||||||||||||||||||||||||||||||||
|
Draw off the supernatant butanolic solution with the aid of a pipette, and transfer it to a centrifuge tube (4.6) and centrifuge. Then measure the optical density EA with a spectrophotometer at 555 nm using n-butanol (3.12) as blank. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Control test |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a control test, using the same procedure, starting from point 5.2, on 20.0 ml of diluted methanol (3.2). Measure the optical density EB at 555 nm using n-butanol (3.12) as blank. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6 Standard test |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a standard test, using the same procedure, starting from point 5.2, on 20.0 ml of standard solution (3.14.2). Measure the optical density EC at 555 nm using n-butanol (3.12) as blank. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The ethopabate content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
(EA - EB) x 80 |
||||||||||||||||||||||||||||||||||||||||||||
|
___________ |
||||||||||||||||||||||||||||||||||||||||||||
|
(EC - EB) x W |
||||||||||||||||||||||||||||||||||||||||||||
|
in which |
||||||||||||||||||||||||||||||||||||||||||||
|
--EA = optical density of the solution from the sample; |
||||||||||||||||||||||||||||||||||||||||||||
|
--EB = optical density of the solution resulting from the control test; |
||||||||||||||||||||||||||||||||||||||||||||
|
--EC = optical density of the solution resulting from the standard test; |
||||||||||||||||||||||||||||||||||||||||||||
|
--W = weight of test portion in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
20%, relative to the higher result, for contents of ethopabate below 7.5 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
1.5 mg/kg in absolute value, for contents between 7.5--10 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
15%, relative to the higher result, for contents above 10 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
10. DETERMINATION OF DINITOLMIDE (DOT) |
||||||||||||||||||||||||||||||||||||||||||||
|
(3, 5-dinitro-o-toluamide) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of dinitolmide (DOT) in feeding stuffs, concentrates and premixtures. Nitrofuran derivatives may interfere. The lower limit of the determination is 40 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with acetonitrile. The extract is purified on aluminium oxide and filtered. An aliquot of the filtrate is evaporated to dryness. The residue is dissolved in dimethylformamide and treated with ethylenediamine forming a purple complex. Dinitolmide is determined by spectrophotometry at 560 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Acetonitrile 85% (v/v): mix 850 ml of pure acetonitrile and 150 ml of water. Before use, distil the mixture and collect the fraction which boils between 75--77°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Aluminium oxide for column chromatography: heat at 750°C for at least 2 hours, cool in desiccator and keep in a brown glass bottle with a ground glass stopper. Before use humidify as follows: place in a brown glass bottle 10 g of aluminium oxide and 0.7 ml of water, stopper, heat for 5 minutes in a bath of boiling water, shaking vigorously, let it cool, still shaking. Check strength by subjecting to analysis, starting from point 5.1, a determined quantity of standard solution (3.6). The recovery rate of the dinitolmide must be 100%±2%. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 N, N-dimethylformamide 95% (v/v): mix 95.0 ml of N, N-dimethylformamide and 5.0 ml of water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Diaminoethane, maximum water content: 2.0%. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Standard substance: pure 3, 5-dinitro-o-toluamide complying with the characteristics below: |
||||||||||||||||||||||||||||||||||||||||||||
|
melting point: 177°C; |
||||||||||||||||||||||||||||||||||||||||||||
|
molecular extinction coefficient at 248 nm in acetonitrile: 13.1 x 103 |
||||||||||||||||||||||||||||||||||||||||||||
|
molecular extinction coefficient at 266 nm in N, N-dimethylformamide: 10.1 x 103. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Standard solution: weigh, to the nearest 0.1 mg, 40 mg of standard substance (3.5), dissolve with acetonitrile (3.1) in a 200 ml volumetric flask, make up to volume with the same solvent and mix. Dilute 20.0 ml to 100 ml with acetonitrile (3.1) in a volumetric flask and mix. 1 ml of this solution contains 40 µg of dinitolmide. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flask with ground glass stopper. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Reflux condenser with ground glass joint. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Filtering crucible porosity G3, diameter 60 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Vacuum filter (such as Witt apparatus). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Water bath, set at 50°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Spectrophotometer with suitable cells (10 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Extraction and purification |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 10 g of the finely divided and mixed sample. For concentrates and premixtures, weigh approximately 1 g, to the nearest mg. Place the test portion in a conical flask (4.1) and add 65 ml of acetonitrile (3.1). Mix, fit reflux condenser (4.2) to the flask and heat in the water bath (4.5) for 30 minutes, shaking continuously. Cool under stream of cold water. Add 20 g of aluminium oxide (3.2), shake for 3 minutes, leave to settle. |
||||||||||||||||||||||||||||||||||||||||||||
|
Place a 100 ml volumetric flask in the vacuum filter (4.4), fit filtering crucible (4.3) and filter the solution, using suction. Then transfer the remaining solids into the crucible with the aid of a few ml of acetonitrile (3.1) and suck the residue dry. Release the vacuum, suspend the residue again in a few ml of acetonitrile (3.1) and again apply vacuum. Repeat these last operations until the volume of the filtrate reaches about 95 ml. Make up to 100 ml with acetonitrile (3.1) and mix. If necessary, dilute an aliquot with acetonitrile (3.1) to obtain a solution containing 5--15 µg of dinitolmide per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Development of color and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette into three beakers A, B and C respectively, 4.0 ml of the solution obtained in 5.1. Also add to beaker C only 1.0 ml of standard solution (3.6). Place the three beakers on the water bath (4.5), placed under a well ventilated hood, and evaporate until dry in a current of dry air. Cool the three beakers to room temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 10.0 ml of N, N-dimethylformamide (3.3) in beaker A and 2.0 ml in beakers B and C respectively, leave in contact for a few minutes, stirring a little, until the residue completely dissolves. Then add 8.0 ml of diaminoethane (3.4) in beakers B and C and mix. Exactly 5 minutes after adding the diaminoethane measure the optical density of the three solutions in the spectrophotometer (4.6) at 560 nm using the N, N-dimethylformamide (3.3) as a blank. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The dinitolmide content in mg per kg of sample is given by the formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value, for contents of dinitolmide below 100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100--5,000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value, for contents between 5,000--10,000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10,000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
11. DETERMINATION OF NICARBAZIN. |
||||||||||||||||||||||||||||||||||||||||||||
|
(equimolecular mixture of 4, 4-dinitrocarbanilide and 2-hydroxy-4, 6-dimethylpyrimidine). |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of nicarbazin in feeding stuffs, concentrates and premixtures containing not more than 5% grassmeal. Nitrofuran derivatives, acetylenheptine and carbadox may interfere. The lower limit of the determination is 20 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with N, N-dimethylformamide. The extract is purified by chromatography on a column of aluminium oxide; the nicabazine is eluted with ethanol. The eluate is treated with ethanolic sodium hydroxide, forming a yellow colour. Nicarbazin is determined by spectrophotometry at 430 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 N, N-dimethylformamide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Aluminium oxide for column chromatography: heat at 750°C for at least 2 hours, cool in desiccator and keep in a brown glass bottle with a ground glass stopper. Before use, check strength by subjecting to analysis, starting from point 5.2, a determined quantity of standard solution (3.8.3). The recovery rate of the nicarbazin must be 100% ± 2%. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Ethanol solution, 95% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Ethanol solution, 80% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Sodium hydroxide solution, 50% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Ethanolic sodium hydroxide solution, 1% (w/v): put 1 ml of sodium hydroxide solution (3.5) in a 50 ml volumetric flask; make up the volume with 80% ethanol (3.4). Prepare at the time of use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Standard substance: pure nicarbazine, moleucular extinction coefficient at 362 nm in N, N-dimethylformamide: 37.8x103. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Standard solutions: |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8.1 Solution of 1.25 mg of nicarbazin per ml: weigh out to within 0.1 mg, 125 mg of standard substance (3.7). Dissolve in 75 ml of N, N-dimethylformamide (3.1) in a 100 ml volumetric flask, heating slightly. Allow to cool, make up the volume with the same solvent and mix. Keep away from light. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8.2 Solution of 0.125 mg of nicarbazin per ml: dilute 10.0 ml of the solution (3.8.1), to 100 ml with N, N-dimethylformamide (3.1) in a volumetric flask and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8.3 Solution of 0.025 mg of nicarbazin per ml: dilute 20.0 ml of the solution (3.8.2), to 100 ml with N, N-dimethylformamide (3.1) in a volumetric flask and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flask with ground glass stopper. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Reflux condenser with ground glass joint. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Boiling water bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Centrifuge. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Glass tube for chromatography (interior diameter: approximately 25 mm, length: approximately 300 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Spectrophotometer with suitable cells (10 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Burette marked in 1/10th ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 10 g of the finely divided and mixed sample. For concentrates and premixtures, weigh approximately 1 g, to the nearest mg. Place the test portion in a conical flask (4.1) and add exactly 100 ml of N, N-dimethylformamide (3.1). Mix, fit reflux condenser (4.2) on the flask and heat on the water bath (4.3) for 15 minutes, shaking from time to time. Cool under a stream of cold water. Then pour the supernatant layer into a centrifuge tube (4.4) and centrifuge for about 3 minutes. If necessary, dilute 25.0 ml of the supernatant layer with N, N-dimethylformamide (3.1), to obtain a solution containing 2.0--10 µg of nicarbazin per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Run into a tube for chromatography (4.5) a slurry of 30 g of aluminium oxide (3.2) in N, N-dimethylformamide (3.1). Let the liquid level fall to 1 cm above the column of aluminium oxide and then put into the column 25.0 ml of the extract obtained in 5.1. Allow the liquid to flow through, not letting the column get dry, and wash the column with three 10 ml portions of N, N-dimethylformamide (3.1). Then elute with 70 ml of ethanol (3.3). Eliminate the first 10 ml of the eluate and collect the rest, dividing it up as follows: |
||||||||||||||||||||||||||||||||||||||||||||
|
one 5 ml portion (a); |
||||||||||||||||||||||||||||||||||||||||||||
|
one 50 ml portion (b) in a volumetric flask; |
||||||||||||||||||||||||||||||||||||||||||||
|
one 5 ml portion (c). |
||||||||||||||||||||||||||||||||||||||||||||
|
Check that portions (a) and (c) do not turn yellow when ethanolic sodium hydroxide (3.6) is added. Continue the operations on portion (b) as shown in 5.3. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Development of colour and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette 20.0 ml of portion (b) of the eluate in two separate 25 ml volumetric flasks A and B. Add to flask A 5.0 ml of ethanolic sodium hydroxide (3.6) and to flask B 5.0 ml of ethanol (3.3). Mix well. |
||||||||||||||||||||||||||||||||||||||||||||
|
Within the next 5 minutes measure the optical density of both solutions at 430 nm, using a mixture of 20.0 ml of ethanol (3.3) and 5.0 ml of ethanol solution of sodium hydroxide (3.6) as a blank. |
||||||||||||||||||||||||||||||||||||||||||||
|
Subtract the value of the optical density of solution B from that of solution A. From this value determine the quantity of nicarbazin referring to the calibration curve (5.4). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Subject 25.0 ml of the standard solution (3.8.3) to chromatography as shown in 5.2. Transfer 2.0, 4.0, 6.0, 8.0 and 10.0 ml (corresponding to 0.025, 0.050, 0.075, 0.100 and 0.125 mg of nicarbazin respectively) of portion (b) into 25 ml graduated flasks from a burette (4.7). To each flask add 5.0 ml of ethanolic sodium hydroxide (3.6), make the volume up with ethanol (3.3) and mix well. |
||||||||||||||||||||||||||||||||||||||||||||
|
Within the next 5 minutes measure the optical density of the solutions at 430 nm, using a mixture of 20.0 ml of ethanol (3.3) and 5.0 ml of ethanolic sodium hydroxide (3.6) as a control. Trace the calibration curve, using the optical density values as the ordinates and the corresponding quantities of nicarbazin in mg as the abscissae. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The nicarbazin content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity of nicarbazin in mg as determined by photometric measurement; |
||||||||||||||||||||||||||||||||||||||||||||
|
W = weight of test portion in grams; |
||||||||||||||||||||||||||||||||||||||||||||
|
F = coefficient of dilution (see 5.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value, for contents of nicarbazin below 100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100 -- 5 000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value, for contents between 5 000 -- 10 000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10 000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
12 DETERMINATION OF VITAMIN A (RETINOL). |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of Vitamin A in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 10 000 IU/kg for highly pigmented feeds and 4 000 IU/kg for others1. Products are classified in two groups, according to their presumed Vitamin A contents: |
||||||||||||||||||||||||||||||||||||||||||||
|
Group A: contents lower than 200 000 IU/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
Group B: contents equal to or greater than 200 000 IU/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is hydrolised in hot ethanolic potassium hydroxide solution and in the presence of an antioxidant or in a nitrogen atmosphere. The mixture is extracted with 1, 2-dichlorethane. The extract is evaporated to dryness and treated with light petroleum. The solution is chromatographed on a column of aluminium oxide (for Group B products, chromatography is only required in certain cases). For Group A products the Vitamin A is determined by spectrophotometry at 610 nm after development of a coloured complex according to the Carr-Price reaction; for Group B products by spectrophotometry in the UV at 325 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
(1) 1 IU = 0.3 g of Vitamin A. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
( a ) used for analysing products of Groups A and B. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Ethanol, 96% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Sodium ascorbate solution, 10% (w/v) or |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Purified nitrogen. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Potassium hydroxide solution, 50% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Potassium hydroxide solution, 1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Potassium hydroxide solution, 0.5 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 1, 2-dichlorethane. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Light petroleum, boiling range: 40--60°C: if necessary, purify as follows: stir 1 000 ml light petroleum with 20 ml lots of concentrated sulphuric acid until the acid remains colourless. Remove the acid and wash the light petroleum successively with 500 ml water, twice with 250 ml of 10% (w/v) sodium hydroxide solution and three times with 500 ml water. Remove the aqueous layer, dry the light petroleum for 1 hour over active carbon and anhydrous sodium sulphate, filter and distil. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Aluminium oxide, standardised according to Brockmann: ash for 8 hours at 750°C, cool in a desiccator and keep in a brown glass bottle fitted with a ground glass stopper. Before use in chromatography moisten as follows: place in a brown glass bottle 10 g aluminium oxide and 0.9 ml water, seal with a stopper, reheat for 5 minutes in a boiling water bath while shaking. Leave to cool. Verify the activity of the aluminium thus prepared by subjecting a known quantity of Vitamin A (3.17) (approximately 500 IU) to the procedure of 5.3 and 5.4 and checking recovery. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Basic aluminium oxide, degree of activity 1 (Woelm, Merck or equivalent). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Diethyl ether: remove peroxides and traces of water by chromatography on a column of basic aluminium oxide (3.10). (25 g aluminium oxide per 250 ml diethyl ether). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Light petroleum solutions (3.8) containing 4, 8, 12, 16 and 20% (v/v) diethyl ether (3.11). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Sodium sulphide solution 0.5 molar in 70% (v/v) glycerine, prepared from sodium sulphide. |
||||||||||||||||||||||||||||||||||||||||||||
|
( b ) used exclusively for analysing Group A products. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Crystallizable benzene. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.15 Chloroform: remove the ethanol, phosgene and traces of water by chromatography on a column of basic aluminium oxide (3.10) (50 g aluminium oxide per 200 ml chloroform; it is advisable to chromatograph the first 50 ml of the eluate a second time). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.16 Carr-Price reagent: stir approximately 25 g antimony trichloride (kept in a desiccator) with 100 ml chloroform (3.15) until the solution is saturated. A slight deposit of antimony trichloride causes no problem. Add 2 ml acetic anhydride. Keep in a refrigerator in a brown glass bottle with ground glass stopper. The solution keeps for several weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.17 Vitamin A -- standardized spectrophotometrically. |
||||||||||||||||||||||||||||||||||||||||||||
|
( c ) used exclusively for analysing Group B products. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18 Isopropanol, for chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Water bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Vacuum evaporation apparatus with round flasks of different capacities. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Glass chromatography tubes (length: approximately 300 mm; internal diameter: approximately 13 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Spectrophotometer. Measurements in the UV require silica cells. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 UV lamps suitable for 364 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
NB All operations must be carried out away from direct light, if necessary in brown glass equipment. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Test Sample |
||||||||||||||||||||||||||||||||||||||||||||
|
From the finely divided sample, take a test sample proportional to the presumed Vitamin A content, thus: |
||||||||||||||||||||||||||||||||||||||||||||
|
0.1-- 1.0 g for concentrates (contents greater than 20 000 IU/g); |
||||||||||||||||||||||||||||||||||||||||||||
|
3.0 -- 5.0 g for premixtures (contents of between 400 -- 20 000 IU/g); |
||||||||||||||||||||||||||||||||||||||||||||
|
10 -- 20 g for mineral mixtures; |
||||||||||||||||||||||||||||||||||||||||||||
|
30 g for Group A products. |
||||||||||||||||||||||||||||||||||||||||||||
|
Immediately place the test sample in a flask with a ground glass stopper. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Hydrolysis and extraction1 |
||||||||||||||||||||||||||||||||||||||||||||
|
Add successively to the test sample 40 ml ethanol (3.1), 2 ml sodium ascorbate solution (3.2)2, 10 ml potassium hydroxide solution (3.4) and 2 ml sodium sulphide solution (3.13). |
||||||||||||||||||||||||||||||||||||||||||||
|
Heat for 30 minutes at 70-80°C under a reflux condenser and then leave to cool under a stream of water. Add 50 ml ethanol (3.1) and 100 ml 1, 2-dichlorethane (3.7) (taken with a pipette). Shake vigorously and then decant the supernatant liquid into a decanting container. Add to the container 150 ml potassium hydroxide solution (3.5), shake for 30 seconds and leave to stand until the layers are separated. Collect the dichlorethane layer (lower layer) in a decanting container, add 40 ml potassium hydroxide solution (3.6), shake for 10 seconds and leave to stand until the layers are separated. Collect the dichlorethane layer in a decanting container and wash 6-8 times with 40 ml lots of water until free of alkali (phenolphthalein test). Collect the dichlorethane layer and remove the last traces of water using strips of filter paper. |
||||||||||||||||||||||||||||||||||||||||||||
|
Evaporate to dryness an aliquot part of the solution under vacuum and on the water bath at 40°C. Rapidly treat the residue with 5 ml light petroleum (3.8). |
||||||||||||||||||||||||||||||||||||||||||||
|
For Group A products, chromatograph as shown in 5.3.1. For Group B products, transfer the solution to a 50 ml graduated flask, make up to volume with light petroleum (3.8), mix and measure the optical density as shown in 5.4.2. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3.1 Group A products |
||||||||||||||||||||||||||||||||||||||||||||
|
Fill a chromatography tube (4.3) to a height of 200 mm with alumium oxide (3.9) previously slurried with light petroleum (3.8). Place in the tube the solution obtained in 5.2 and immediately add 20 ml light petroleum (3.8). Elute successively with 10 ml lots of the light petroleum solutions at 4, 8, 12, 16 and 20% diethyl ether (3.12) under pressure or partial vacuum, the rate of flow being 2-3 drops per second. |
||||||||||||||||||||||||||||||||||||||||||||
|
The carotene is eluted first3. The Vitamin A is generally eluted with the light petroleum solution at 20% diethyl ether (3.12). The elution is followed under UV light (brief irradiation of the column with the mercury lamp). The fluorescent zone of the Vitamin A is clearly separated from the yellow xanthophyll zones following it. Collect the eluate fraction containing the Vitamin A in an Erlenmeyer flask. |
||||||||||||||||||||||||||||||||||||||||||||
|
1 For milk feeds and products with a tendency to agglomerate or swell double the quantity of the reagents shown in the first and second paragraphs of 5.2. |
||||||||||||||||||||||||||||||||||||||||||||
|
2 Sodium ascorbate need not be added when hydrolysis is carried out in a nitrogen atmosphere. |
||||||||||||||||||||||||||||||||||||||||||||
|
3 Carotene content may be determined by optical density measurement at 450 nm: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
5.3.2 Group B products |
||||||||||||||||||||||||||||||||||||||||||||
|
Chromatography must only be carried out if the optical density measurements obtained in 5.4.2 do not conform to the requirements given in 5.4.2. |
||||||||||||||||||||||||||||||||||||||||||||
|
If chromatography proves necessary, place in the chromatography column an aliquot part of the solution in the light petroleum obtained in 5.2, containing approximately 500 IU of Vitamin A, and chromatograph as shown in 5.3.1. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.1 Group A products |
||||||||||||||||||||||||||||||||||||||||||||
|
Evaporate to dryness under vacuum the eluate containing the Vitamin A obtained in 5.3.1. Treat the residue with 2 ml benzene (3.14). Take 0.3 ml of this solution and add 3 ml of the Carr-Price reagent (3.16). A blue colouring develops. Measure the optical density with the spectrophotometer at 610 nm exactly 30 seconds after the reaction has begun. Determine the Vitamin A content by reference to a standard curve obtained from benzene solutions of increasing Vitamin A-standard concentrations treated with Carr-Price reagent (2-16 IU Vitamin A-standard (3.17) per 0.3 ml benzene (3.14) + 3 ml Carr-Price reagent (3.16). The standard curve must be checked regularly and frequently using the standard and a freshly prepared Carr-Price reagent solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.2 Group B products |
||||||||||||||||||||||||||||||||||||||||||||
|
Take an aliquot part of the solution in light petroleum obtained in 5.2 containing approximately 200 IU Vitamin A. Evaporate to dryness under vacuum and treat the residue with 25 ml isopropanol (3.18). Measure the optical density in the spectrophotometer at 325, 310 and 334 nm. The absorption maximum is located at 325 nm. The Vitamin A content of the solution is calculated as follows: |
||||||||||||||||||||||||||||||||||||||||||||
|
E325 . 18.30 = IU of Vitamin A/ml |
||||||||||||||||||||||||||||||||||||||||||||
|
However, the ratio of the optical densities |
||||||||||||||||||||||||||||||||||||||||||||
|
E310 : E325 and E334: E325 |
||||||||||||||||||||||||||||||||||||||||||||
|
must be 6 : 7 = 0.857. |
||||||||||||||||||||||||||||||||||||||||||||
|
If one of these ratios differs appreciably from this value (< 0.830 or > 0.880), the measurement of the optical densities must be preceded by chromatography in accordance with the method given in 5.3.2. If the measurement of the optical densities carried out after chromatography shows that the above mentioned ratios still differ appreciably from the value of 0.857 (< 0.830 > 0.880), the determination must be carried out in accordance with the method given for Group A products. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the Vitamin A content of the sample taking into account the weight of the test sample and the dilutions carried out in the course of analysis. Express the results in IU of Vitamin A per kg of feeding stuff, concentrate or premixture. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
20%, relative to the higher result, for Vitamin A contents lower than 75,000 IU/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
15 000 IU for contents between 75 000 -- 150 000 IU/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 150 000 -- 250 000 IU/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
25 000 IU for contents between 250 000 -- 500 000 IU/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents greater than 500 000 IU/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
13. DETERMINATION OF THIAMINE (VITAMIN B1, ANEURINE) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of thiamine (aneurine, Vitamin B1 ) in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 5 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The solution is treated when hot with dilute sulphuric acid and then hydrolysed enzymatically. The solution obtained is subjected to alkaline oxidation. The thiochrome formed is extracted with isobutanol and determined by fluorimetry. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 100 µg/ml standard thiamine solution: dissolve 127.1 mg pure thiaminium dichloride, previously dried under vacuum to constant weight, or 113.4 mg pure thiaminium chloride, treated in the same fashion, in 1000 ml of sulphuric acid 0.2 N (3.2). If stored in a cool, dark place, this solution keeps for 1 month. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Sulphuric acid 0.2 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Sodium bisulphite. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Potassium ferricyanide solution, 20% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Potassium hydroxide solution, 25% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Oxidizing mixtures: mix 2 ml potassium ferricyanide solution (3.4) with 48 ml potassium hydroxide solution (3.5). This mixture does not keep for more than 4 hours. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Isobutanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Sodium acetate solution, 2.5 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Multienzymatic preparation containing protease, phosphatase and amylase (e.g. Clarase). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Ethanol, 96% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Water bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Centrifuge (3 500 rpm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Fluorimeter. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Enzymatic hydrolysis |
||||||||||||||||||||||||||||||||||||||||||||
|
Place in each of two 250 ml graduated flasks, A and B, identical amounts of the finely divided sample containing approximately 100 µg thiamine and 125 ml sulphuric acid (3.2). Also add, to flask A only, 1.0 ml standard solution (3.1) (internal standard). Shake the flasks vigorously, place on a boiling water bath and keep there for 15 minutes, shaking occasionally. Leave to cool to approximately 45°C. Add to each flask 20 ml sodium acetate solution (3.8) and 0.5 g multienzymatic preparation (3.9), then leave to stand for 20 minutes at room temperature. Add 20 ml sodium acetate solution (3.8), make up to volume with water, mix and filter. Collect filtrates A and B after having discarded the first 15 ml. Prepare the following solutions: |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1.1 Reference solution T |
||||||||||||||||||||||||||||||||||||||||||||
|
Place in a centrifuge tube (4.2) 5 ml filtrate A and approximately 10 mg sodium bisulphite (3.3). Immerse the tube in a boiling water bath for 15 minutes and then leave to cool to room temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1.2 Solutions A (internal standard) and B (sample). |
||||||||||||||||||||||||||||||||||||||||||||
|
Place 5 ml filtrate A in a centrifuge tube (4.2) and 5 ml filtrate B in another centrifuge tube (4.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Oxidation |
||||||||||||||||||||||||||||||||||||||||||||
|
Add to solutions T, A and B 5 ml of the oxidising mixture (3.6) and, one minute later 10 ml isobutanol (3.7). Stopper the tubes and shake vigorously for 5 seconds. Leave to stand for 1 minute and centrifuge so as to separate the layers. From each tube transfer 5 ml of the supernatant isobutanol layer to each of the 25 ml graduated flasks, make up to volume with ethanol (3.10) and mix (= extracts T, A and B). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Measurement of fluorescence |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out the measurements at the wavelength for which the fluorimeter gives an optimal response to the fluorescence of the thiochrome. Irradiate at approximately 365 nm. Adjust the instrument to zero using extract T. Measure the intensity of fluorescence of extracts A and B. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The Thiamine content in mg/kg of the sample is given by the formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
where: |
||||||||||||||||||||||||||||||||||||||||||||
|
a = intensity of fluorescence of extract A (internal standard); |
||||||||||||||||||||||||||||||||||||||||||||
|
b = intensity of fluorescence of extract B (sample); |
||||||||||||||||||||||||||||||||||||||||||||
|
c = weight of the test sample in grams; |
||||||||||||||||||||||||||||||||||||||||||||
|
d = amount of thiamine in µg added to the test sample (internal standard). |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents lower than 500 mg/kg, and 5%, relative to the higher result, for contents equal to or greater than 500 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
14. DETERMINATION OF ASCORBIC ACID AND DEHYDROASCORBIC ACID (VITAMIN C) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the total content of ascorbic and dehydroascorbic acids (Vitamin C) in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 5 mg/kg. Products are classified in two groups, according to their presumed Vitamin C content. |
||||||||||||||||||||||||||||||||||||||||||||
|
Group A: contents lower than 10 g/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
Group B: contents equal to or greater than 10 g/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is suspended in a dilute solution of metaphosphoric acid and extracted with chloroform. The aqueous phase is treated with a solution of 2, 6-dichlorophenol-indophenol in order to transform the ascorbic acid into dehydroascorbic acid, and then with a solution of 2, 4-dinitrophenylhydrazine. The hydrazone formed is extracted with a mixture of ethyl acetate, glacial acetic acid and acetone. The solution is chromatographed on a column of silica gel, the eluate evaporated to dryness and the residue dissolved in dilute sulphuric acid. The optical density of the solution is measured by a spectrophotometer at 509 nm. For Group A products the eluate resulting from chromatography on the column is further subjected to thin layer chromatography to isolate the hydrazone. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 L-ascorbic acid solution, 0.05%: dissolve 50 mg L-ascorbic acid in approximately 20 ml metaphosphoric acid solution (3.2) and make up to 100 ml with water. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Metaphosphoric acid solution 10% (w/v): after grinding it in a mortar, dissolve in water 200 g metaphosphoric acid and make up to 2000 ml with water. Keep at 4°C. This is stable for 1 week. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Chloroform. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 2, 6-dichlorophenol-indophenol solution, 0.5% (w/v). Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Filtration aid (S. and S. No. 121 or equivalent). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 2, 4-dinitrophenylhydrazine solution, 2% (w/v): dissolve 2 g 2, 4-dinitrophenylhydrazine in 100 ml dilute sulphuric acid (25 ml sulphuric acid, d: 1.84, diluted by making up to 100 ml with water). Stored at a cool temperature this solution keeps for 1 week. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Nitrogen, or |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Carbon dioxide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Mixture of ethyl acetate/glacial acetic acid/acetone: 96/2/2 in volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Mixture of dichloromethane/glacial acetic acid: 97/3 in volume. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Silica gel, particle size: 0.05--0.2 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Stahl grade silica gel H, for thin layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Dilute sulphuric acid: place 105 ml water in a 200 ml graduated flask, make up to volume with sulphuric acid, d: 1.84. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Eluting solvent for thin layer chromatography: mix 75 ml diethyl ether, 25 ml ethyl acetate and 4.0 ml 96% (w/v) acetic acid. Renew after 2--3 chromatographs. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Water bath fitted with a thermostat set at 20°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Centrifuge (3 500 rpm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Rotary vacuum evaporator. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Glass chromatography tubes (length: approximately 100 mm, internal diameter: approximately 20 mm), with a sintered disc (e.g. Allihn tubes). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Spectrophotometer or colorimeter with filters. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Apparatus for thin layer chromatography, with silica gel plates (3.12) coated to a depth of 0.5--0.6 mm. (Ready-made plates are appropriate). Dry the plates for 2½--3 hours in the drying oven at 120--130°C. Leave to cool and then keep in a desiccator for at least 24 hours before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Drying oven set at 120--130°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Place in each of two 250 ml graduated flasks (with ground glass stoppers), A and B, identical quantities of the finely divided sample containing about 200 µg vitamin C. Add to flask A only 0.4 ml standard solution (3.1) and mix, shaking gently (internal standard). |
||||||||||||||||||||||||||||||||||||||||||||
|
Add to each flask 30 ml chloroform (3.3) and 25 ml metaphosphoric acid solution (3.2) at 4°C. Shake briefly and then leave to stand for 10--15 minutes. Add 25 ml water, stopper the flasks, shake vigorously for 10 seconds and leave to stand for 10--15 minutes in the water bath (4.1). Centrifuge to separate the aqueous phase from the chloroform phase. Carry out the operations simultaneously, as described below, on the aqueous extracts A (internal standard) and B. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Oxidation |
||||||||||||||||||||||||||||||||||||||||||||
|
Using a pipette, transfer 40 ml of the supernatant aqueous solution (slightly cloudy) obtained in 5.1 to a reaction tube fitted with a ground glass stopper, add 0.5--1 ml 2, 6-dichlorophenol-indophenol solution (3.4) and mix. A red colouring develops which should remain for at least 15 minutes. Then add approximately 300 mg filtration aid (3.5), shake and filter through a dry pleated filter. The filtrate need not necessarily be clear. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Reaction with 2, 4-dinitrophenylhydrazine and hydrazone extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Using a pipette, transfer 10 ml of the filtrate obtained in 5.2 to a centrifuge tube (4.2), add 2 ml 2, 4-dinitrophenylhydrazine solution (3.6) and mix. Pass a stream of nitrogen (3.7) or carbon dioxide (3.8) rapidly into the tube. stopper the tube and immerse it for approximately 15 hours (overnight) in the water bath (4.1). Then add 3 ml water, 20 ml of the ethyl acetate/glacial acetic acid/acetone mixture (3.9) and approximately 800 mg filtration aid (3.5). Stopper the tube, shake vigorously for 30 seconds and centrifuge. Place 15 ml of the supernatant phase in an evaporation flask and evaporate under reduced pressure in the rotary evaporator (4.3) until an oily residue is obtained. Dissolve the residue in 2 ml of the ethyl acetate/glacial acetic acid/acetone mixture (3.9) by reheating at 50°C, leave to cool, add 10 ml of the dichloromethane/glacial acetic acid mixture (3.10) and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Chromatography on a column |
||||||||||||||||||||||||||||||||||||||||||||
|
Fill a chromatography tube (4.4) up to a level of 30 mm with the dichloromethane/glacial acetic acid mixture (3.10). Suspend (shaking vigorously) 5 g silica gel (3.11) in 30 ml of the dichloromethane/glacial acetic acid mixture (3.10); pour the suspension into the tube. Leave to stand and then compress under nitrogen (3.7) at low pressure. Decant into the tube the solution obtained in 5.3, rinse the flask with a small quantity of the dichloromethane/glacial acetic acid mixture (3.10) and decant into the tube, then fill the latter with the mixture (3.10) and proceed to wash the column with the same mixture (3--4 lots of approximately 5 ml) until a colourless eluate is obtained. Discard the part of the eluate which is coloured yellow. Elute the reddish zone at the top of the column with the ethyl acetate/glacial acetic acid/acetone mixture (3.9), collect the eluate and evaporate to dryness. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.1 For Group A products (contents in vitamin C lower than 10 g/kg), dissolve the residue in 2.0 ml of the ethyl acetate/glacial acetic acid/acetone mixture (3.9) and chromatograph immediately on a thin layer as shown in 5.5. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.2 For Group B products (contents in vitamin C equal to or greater than 10 g/kg), treat the oily residue with 4.0 ml dilute sulphuric acid (3.13), shake vigorously to dissolve the residue completely and measure the optical density as shown in 5.6. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Thin layer chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out in duplicate the operations described as follows. Place in a thin line on the plate (4.6) 0.5 ml of the solution obtained in 5.4.1. Using the eluting solvent (3.14) develop for at least 20 minutes in a tank saturated with solvent vapour, until the pink-coloured hydrazone zone is clearly separated. Leave to dry in the open. Mark out the limit of the pink zone, scrape away the zone with a spatula and quantitatively transfer the powder into a chromatography tube (4.4). |
||||||||||||||||||||||||||||||||||||||||||||
|
Elute successively once with 2 ml and twice with 1.5 ml of the ethyl acetate/glacial acetic acid/acetone mixture (3.9). Collect the eluate in a small flask (the last part must be colourless). Evaporate to dryness, treat the oily residue with 4.0 ml dilute sulphuric acid (3.13), shake vigorously to dissolve the residue completely and measure the optical density. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6 Measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the optical density with a spectrophotometer at 509 nm 20--30 minutes after dissolving the residue in sulphuric acid. Carry out the measurements by comparison with dilute sulphuric acid (3.13). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.7 Blank test |
||||||||||||||||||||||||||||||||||||||||||||
|
Carry out a blank test applying the same procedure but without the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The vitamin C content of the sample in g per kg is given by the formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
where: |
||||||||||||||||||||||||||||||||||||||||||||
|
a = optical density of the blank; |
||||||||||||||||||||||||||||||||||||||||||||
|
b = optical density of the internal standard solution; |
||||||||||||||||||||||||||||||||||||||||||||
|
c = optical density of the sample solution; |
||||||||||||||||||||||||||||||||||||||||||||
|
d = weight, in grams, of the test sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for vitamin C contents lower than 10 g/kg, and |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents equal to or greater than 10 g/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
15. DETERMINATION OF MENADIONE (VITAMIN K3) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the quantity of menadione (vitamin K3) in feeding stuffs, concentrates and premixtures. The lower limit of the determination is 1mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with diluted ethanol. The mixture is clarified with tannin solution and centrifuged. The extract is treated with a solution of sodium carbonate; the liberated menadione is extracted with 1, 2-dichloroethane. The dichloroethane extract is treated, according to its menadione content, either directly or after evaporation, with 2, 4-dinitrophenylhydrazine in solution in ethanol acidified with hydrochloric acid. The obtained hydrazone treated with ammonia in excess gives rise to a blue-green coloured complex the optical density of which is measured at 635 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Ethanol, 96% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Ethanol (3.1) diluted to 40% with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Tannin solution, 10% (w/v), prepared from purified powdered tannin. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 1, 2-dichloroethane. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Anhydrous sodium carbonate solution, 10% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Hydrochloric acid solution, 37% (w/v), d: 1.19. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Absolute ethanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 2, 4-dinitrophenylhydrazine reagent: dissolve 40 mg 2, 4-dinitrophenylhydrazine in about 40 ml boiling absolute ethanol (3.7), allow to cool and transfer into a 50 ml volumetric flask. Add 1 ml hydrochloric acid (3.6) and make up to volume with absolute ethanol (3.7). Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Ammonia solution, 25% (w/v), d: 0.91. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Ammoniacal ethanol: mix one volume of ethanol (3.7) with one volume of ammonia (3.9). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Standard solutions of menadione: dissolve 20 mg menadione (vitamin K3) in 1, 2-dichloroethane (3.4) and make up to 200 ml. Dilute aliquots of this stock solution with 1, 2-dichloroethane (3.4) to obtain a series of solutions with menadione concentrations between 2--10 µg per ml. These solutions must be freshly prepared. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Mechanical shaker. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Centrifuge (3,000--5,000 rpm). |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Separators with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Rotary vacuum evaporator. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Water bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Spectrophotometer. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
NB · All operations must be carried out away from direct light, using apparatus of amber glass where necessary. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Test Sample |
||||||||||||||||||||||||||||||||||||||||||||
|
From the finely divided sample, take a test sample according to the presumed menadione content, e.g.: |
||||||||||||||||||||||||||||||||||||||||||||
|
0.1--5.0 g for concentrates and premixtures; |
||||||||||||||||||||||||||||||||||||||||||||
|
20--30 g for feeding stuffs. |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer the test sample to a flask with ground glass stopper without delay. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Add to the test sample exactly 96 ml dilute ethanol (3.2) and shake mechanically for 15 minutes at room temperature. Then add 4.0 ml tannin solution (3.3), mix, transfer the extract into a centrifuge tube, centrifuge at 3,000--5,000 rpm and decant. Place 20--40 ml, accurately measured, of the extract in a seperator, add by pipette 50 ml 2, 3-dichloroethane (3.4), mix and add by pipette 20 ml sodium carbonate solution (3.5). Shake vigorously for 30 seconds and then collect the dichloroethane phase in a separator (4.3). Add 20 ml water, shake again for 15 seconds, collect the dichloroethane phase and remove traces of water with strips of filter paper. |
||||||||||||||||||||||||||||||||||||||||||||
|
For concentrates and premixtures, take an aliquot part of the extract and dilute with 1, 2-dichloroethane (3.4) to obtain a menadione concentration of 2--10 µg per ml. For feeding stuffs, evaporate to dryness an aliquot part of the extract under reduced pressure in an atmosphere of nitrogen on a water bath at 40°C. Rapidly treat the residue with 1, 2-dichloroethane (3.4) to obtain a solution containing 2--10 µg menadione per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Hydrazone formation |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer 2.0 ml of the dichloroethane extract obtained in 5.2 to a 10 ml volumetric flask and add 3.0 ml 2, 4-dinitrophenylhydrazine reagent (3.8), securely stopper the flask with a cork or teflon or other suitable stopper so as to prevent evaporation, and heat for 2 hours at 70°C on a water bath (4.5). Allow to cool, add 3.0 ml ammoniacal ethanol (3.10), mix, make up to volume with absolute ethanol (3.7) and mix again. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the optical density of the blue-green coloured complex by the spectrophotometer (4.6) at 635 nm by comparison with a reagent blank obtained by treating 2.0 ml 1, 2-dichloroethane (3.4) as indicated in 5.3. |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the quantity of menadione by reference to a calibration curve established for each series of analyses. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Treat 2.0 ml of the menadione standard solutions (3.11) as described in 5.3. Measure the optical density as indicated in 5.4. Plot the calibration curve with the optical density values as ordinates and the corresponding quantities of menadione in µg as) abscissae. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the menadione content of the sample by taking account of the weight of the test sample and of the dilutions carried out in the course of analysis. |
||||||||||||||||||||||||||||||||||||||||||||
|
Express the result in mg menadione per kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
20%, relative to the higher result, for menadione contents less than 10 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
2 mg/kg, in absolute value, for contents between 10--14 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
15%, relative to the higher result, for contents between 14--100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
15 mg/kg, in absolute value, for contents between 100--150 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents greater than 150 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
16. DETERMINATION OF HYDROCYANIC ACID |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of hydrocyanic acid, free and combined in the form of glucosides, in feeding stuffs and in particular in products derived from linseed, manioc flour and certain species of beans. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is suspended in water. The hydrocyanic acid is released by the action of enzymes, entrained by steam distillation and collected in a specific volume of acidified silver nitrate solution. The silver cyanide is separated by filtration and the excess silver nitrate is titrated with a solution of ammonium thiocyanate. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 A suspension of sweet almonds: crush twenty blanched sweet almonds in 100 ml of water at 37-40°C. Check that there is no hydrocyanic acid in 10 ml of the suspension using sodium picrate paper or by carrying out a blank test as described in the last paragraph of 5. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Sodium acetate solution, 10% (w/v) neutral to phenolphthalein. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Anti-foaming emulsion (e.g. silicone). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Nitric acid, d: 1.40. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Silver nitrate solution, 0.02 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Ammonium thiocyanate solution, 0.02 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Ammonium ferric sulphate saturated solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Ammonia, d: 0.958. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Oven with thermostat set at 38°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Apparatus for distillation by entrainment in steam fitted with a condenser with a curved extension piece. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 1000 ml flat-bottomed flasks with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Oil bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Burette graduated in ½0 ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest 5 mg, approximately 20 g of the sample. Place in 1 litre flat-bottomed flask and add 250 ml of water and 10 ml of sweet almond suspension (3.1). Stopper the flask and transfer to the oven for 16 hours at 38°C. Next, cool to room temperature and add 80 ml of water, 10 ml of sodium acetate solution (3.2) and a drop of anti-foaming emulsion (3.3). Connect the flask to the steam distillation apparatus (4.2) and place in an oil bath (4.4) which has first been brought to a temperature slightly above 100°C. Distil 200--300 ml of liquid by passing a powerful current of steam through the flask and gently heating the oil bath (4.4). Collect the distillate in an Erlenmeyer flask protected from the light and containing exactly 50 ml of silver nitrate solution 0.02 N (3.5) and 1 ml of nitric acid (3.4). Make sure that the condenser's delivery adaptor is immersed in the silver nitrate solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer the contents of the Erlenmeyer flask to a 500 ml volumetric flask, make up to volume with water, stir and filter. Remove 250 ml of the filtrate, add approximately 1 ml solution of ammonium ferric sulphate (3.7) and back-titrate the excess silver nitrate with the solution of ammonium thiocyanate 0.02 N (3.6) taken from the burette graduated in ½0 ml (4.5). |
||||||||||||||||||||||||||||||||||||||||||||
|
A blank test may, if required, be carried out by applying the same procedure to 10 ml of sweet almond suspension (3.1), omitting the sample to be analysed. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
If the blank test indicates that silver nitrate solution 0.02 N has been consumed, subtract the value of this from the volume consumed by the distillate of the sample. 1 ml of AgNO3 0.02 N corresponds to 0.54 mg of HCN. Express the result as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Observation |
||||||||||||||||||||||||||||||||||||||||||||
|
If the sample contains a large quantity of sulphides (eg beans), a black precipitate of silver sulphide is formed which is filtered together with the silver cyanide deposit. The formation of this precipitate causes a loss of silver nitrate solution 0.02 N, the volume of which must be subtracted from the volume used to calculate the HCN content. To do this, proceed as follows: |
||||||||||||||||||||||||||||||||||||||||||||
|
Treat the deposit left on the filter with 50 ml of ammonia (3.8) in order to dissolve the silver cyanide. Wash the residue in dilute ammonia and then determine its silver content. Convert the value obtained into ml of silver nitrate solution 0.02 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample's HCN content may also be determined by titrating the acidified ammoniacal filtrate with nitric acid. |
||||||||||||||||||||||||||||||||||||||||||||
|
17. DETERMINATION OF FREE AND TOTAL GOSSYPOL |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the levels of free gossypol, total gossypol and chemically related substances in cottonseed, cottonseed meal and cottonseed cake and in compound feedingstuffs containing these substances where more than 20 mg/kg are present. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The gossypol is extracted in the presence of 3-aminopropan-1-ol, either with a mixture of propan-2-ol and hexane, for the determination of free gossypol, or with dimethylformamide, for the determination of total gossypol. The gossypol is converted by aniline into gossypol-dianiline, the optical density of which is measured at 440 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Propan-2-ol-hexane mixture: mix 60 parts by volume of propan-2-ol with 40 parts by volume of n-hexane. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Solvent A: place in a 1 litre graduated flask approximately 500 ml of propan-2-ol-hexane mixture (3.1), 2 ml of 3-aminopropan-1-ol, 8 ml of glacial acetic acid and 50 ml of water. Make up to volume with the propan-2-ol-hexane mixture (3.1). This reagent is stable for one week. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Solvent B: pipette 2 ml of 3-aminopropan-1-ol and 10 ml of glacial acetic acid into a 100 ml graduated flask. Cool to room temperature and make up to volume with N, N-dimethylformamide. This reagent is stable for one week. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Aniline: if the optical density in the blank test exceeds 0.022, distil the aniline over zinc dust, discarding the first and last 10% fractions of the distillate. Refrigerated and stored in a brown, stoppered glass flask, this reagent will keep for several months. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Standard gossypol solution A: place 27.9 mg of gossypol acetate in a 250 ml graduated flask. Dissolve and make up to volume with solvent A (3.2). Pipette 50 ml of this solution into a 250 ml graduated flask and make up to volume with solvent A. The gossypol concentration of this solution is 0.02 mg per ml. Leave to stand for one hour at room temperature before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Standard gossypol solution B: place 27.9 mg of gossypol acetate in a 50 ml graduated flask. Dissolve and make up to volume with solvent B (3.3). The gossypol concentration of this solution is 0.5 mg per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
Standard gossypol solutions A and B will remain stable for 24 hours if protected from the light. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Shaker: approximately 35 rpm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Spectrophotometer. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Test sample |
||||||||||||||||||||||||||||||||||||||||||||
|
The amount of test sample used depends on the presumed gossypol content of the sample. It is preferable to work with a small test sample and a relatively large aliquot part of the filtrate, so as to obtain sufficient gossypol for precise photometric measurement to be possible. For the determination of free gossypol in cottonseed, cottonseed meal and cottonseed cake, the test sample should not exceed 1 g; for compound feedingstuffs, it may be as much as 5 g. A 10 ml aliquot part of filtrate is suitable in most cases; it should contain 50--100 ug of gossypol. For the determination of total gossypol, the test sample should be between 0.5--5 g, so that a 2 ml aliquot part of filtrate will contain 40--200 µg of gossypol. |
||||||||||||||||||||||||||||||||||||||||||||
|
The analysis should be carried out at a room temperature of about 20°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Determination of free gossypol |
||||||||||||||||||||||||||||||||||||||||||||
|
Place the test sample in a ground-necked 250 ml flask, the bottom of the flask having been covered with crushed glass. Using a pipette, add 50 ml of solvent A (3.2), stopper the flask and mix for one hour in the shaker. Filter through a dry filter and collect the filtrate in a small ground-necked flask. During filtration, cover the funnel with a watch glass. Pipette identical aliquot parts of filtrate containing 50--100 µg of gossypol into each of two 25 ml graduated flasks (A and B). If necessary, make up the volume to 10 ml with solvent A (3.2). Then make the contents of flask (A) up to volume with the propan-2-ol-hexane mixture (3.1). This solution will be used as a reference solution against which to measure the sample solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette 10 ml of solvent A (3.2) into each of two other 25 ml graduated flasks (C and D). Make the contents of flask (C) up to volume with the propan-2-ol-hexane mixture (3.1). This solution will be used as a reference solution against which to measure the blank test solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 2 ml of aniline (3.4) to each of flasks (D) and (B). Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), mix and leave to stand for one hour. |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the optical density of the blank test solution (D) by comparison with the reference solution (C), and the optical density of the sample solution (B) by comparison with the reference solution (A), in the spectrophotometer at 440 nm using 1 cm glass cells. |
||||||||||||||||||||||||||||||||||||||||||||
|
Substract the optical density of the blank test solution from that of the sample solution (= corrected optical density). From this value calculate the free gossypol content as indicated in 6. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Determination of total gossypol |
||||||||||||||||||||||||||||||||||||||||||||
|
Place a test sample containing 1--5 mg of gossypol in a 50 ml graduated flask and add 10 ml of solvent B (3.3). At the same time, prepare a blank test, placing 10 ml of solvent B (3.3) in another 50 ml graduated flask. Heat the two flasks for 30 minutes over a boiling water bath. Cool to room temperature and make the contents of each flask up to volume with the propan-2-ol-hexane mixture (3.1). Mix and leave to settle for 10--15 minutes, then filter and collect the filtrates in ground-necked flasks. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette 2 ml of the sample filtrate into each of two 25 ml graduated flasks, and 2 ml of the blank test filtrate into each of two other 25 ml flasks. Make the contents of one flask from each series up to 25 ml with the propan-2-ol-hexane mixture (3.1). These solutions will be used as reference solutions. |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 2 ml of aniline (3.4) to each of the other two flasks. Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to 25 ml with the propan-2-ol-hexane mixture (3.1), mix and leave to stand for one hour. |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the optical density as indicated in 5.2 for free gossypol. From this value calculate the total gossypol content as indicated in 6. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Results may be calculated either from the specific optical density (6.1), or by reference to a calibration curve (6.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1 From the specific optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
The specific optical densities, under the conditions described, will be the following: |
||||||||||||||||||||||||||||||||||||||||||||
|
free gossypol: |
||||||||||||||||||||||||||||||||||||||||||||
|
total gossypol: |
||||||||||||||||||||||||||||||||||||||||||||
|
The free or total gossypol content of the sample is calculated by using the following formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
where: |
||||||||||||||||||||||||||||||||||||||||||||
|
E = corrected optical density, determined as indicated in 5.2; |
||||||||||||||||||||||||||||||||||||||||||||
|
p = test sample in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
a = aliquot part of the filtrate in ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2 From a calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2.1 Free gossypol |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare 2 series of five 25 ml graduated flasks. Pipette aliquots of 2.0, 4.0, 6.0, 8.0 and 10.0 ml of standard gossypol solution A (3.5) into each series of flasks. Make up the volumes to 10 ml with solvent A (3.2). Complete each series with a 25 ml graduated flask containing only 10 ml of solvent A (3.2) (blank test). |
||||||||||||||||||||||||||||||||||||||||||||
|
Make the volume of the flasks in the first series (including the flask for the blank test) up to 25 ml with the propan-2-ol-hexane mixture (3.1) (reference series). |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 2 ml of aniline (3.4) to each flask in the second series (including the flask for the blank test). Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), mix and leave to stand for one hour (standard series). |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine as indicated in 5.2 the optical density of the solutions in the standard series by comparison with the corresponding solutions in the reference series. Trace the calibration curve by plotting the optical densities against the quantities of gossypol (in µg). |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2.2 Total gossypol |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare six 50 ml graduated flasks. In the first flask place 10 ml of solvent B (3.3), and in the others 2.0, 4.0, 6.0, 8.0 and 10.0 ml of standard gossypol solution B (3.6) respectively. Make the contents of each flask up to 10 ml with solvent B (3.3). Heat for 30 minutes over a boiling water bath. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1) and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
Place 2.0 ml of these solutions in each of two series of six 25 ml graduated flasks. Make the contents of the flasks in the first series up to 25 ml with the propan-2-ol-hexane mixture (3.1) (reference series). |
||||||||||||||||||||||||||||||||||||||||||||
|
Add 2 ml of aniline (3.4) to each flask in the second series. Heat for 30 minutes over a boiling water bath. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), mix and leave to stand for one hour (standard series). |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine as indicated in 5.2 the optical density of the solutions in the standard series by comparison with the corresponding solutions in the reference series. Trace the calibration curve by plotting the optical densities against the quantities of gossypol (in µg). |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results, of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
--15%, relative to the higher result for gossypol contents of less than 500 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
--75 ppm, in absolute value, for contents of not less than 500 ppm and not more than 750 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
--10% relative to the higher result, for contents of more than 750 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
18. DETERMINATION OF THEOBROMINE |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of theobromine in the by-products of the processing of cocoa beans. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The theobromine is extracted with chloroform. The extract is evaporated until dry, dissolved in water and treated with a specific quantity of silver nitrate solution. The nitric acid liberated is titrated with a solution of sodium hydroxide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Chloroform. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Ammonia, d: 0.958. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Sodium sulphate, anhydrous. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Sodium hydroxide solution, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Silver nitrate solution, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Ethanolic solution of phenol red, 1% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Light petroleum, boiling range 40--60°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
Flat-bottomed 500 ml flasks with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, a sample of not more than 10 g containing not more than 80 mg of theobromine, place in a 500 ml flat-bottomed flask with a ground glass stopper and add 270 ml of chloroform (3.1) and 10 ml of ammonia (3.2). Stopper the flask and shake vigorously for 5 minutes. Add 12 g of anhydrous sodium sulphate (3.3), shake again and leave to settle until the following day. Filter into a 500 ml Erlenmeyer and wash the residue with 100 ml of chloroform (3.1). Distil the solvent and eliminate the last traces over a boiling water bath. Redissolve the extract in 50 ml of water and bring to the boil. |
||||||||||||||||||||||||||||||||||||||||||||
|
Cool, neutralise exactly with the sodium hydroxide solution (3.4) using 0.5 ml of phenol red solution (3.6). Add 20 ml of silver nitrate solution (3.5). Titrate the nitric acid liberated with a solution of sodium hydroxide (3.4) until the indicator changes colour (pH 7.4). |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
1 ml of 0.1 N NaOH = 18 mg of theobromine. |
||||||||||||||||||||||||||||||||||||||||||||
|
Express the results as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Observation |
||||||||||||||||||||||||||||||||||||||||||||
|
Products containing more than 8% of crude fatty matter must first be defatted by a 6-hour extraction with light petroleum (3.7). |
||||||||||||||||||||||||||||||||||||||||||||
|
19. DETERMINATION OF VOLATILE MUSTARD OIL |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of mustard oil, separated under steam and expressed as allyl isothiocyanate, in cakes made from the Brassica and Sinapis species, and in compound feeding stuffs which contain cakes made from those species. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is suspended in water. The mustard oil is released by the action of enzymes, separated by distillation with ethanol and collected in dilute ammonia. The solution is treated while warm with a given volume of silver nitrate solution, then cooled and filtered. The excess silver nitrate is titrated with a solution of ammonium thiocyanate. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 White mustard (Sinapis alba). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Ethanol, 95-96% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Anti-foaming emulsion (eg silicone). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Ammonia, d: 0.958. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Silver nitrate solution, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Ammonium thiocyanate solution, 0.1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Nitric acid, d: 1.40. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Ammonium ferric sulphate, saturated solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Flat-bottomed 500 ml flasks with ground glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Distilling apparatus fitted with a condenser and with equipment for preventing the entrainment of droplets. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 10 g of the sample, place in a 500 ml flat-bottomed flask (4.1) and add 2 g of finely ground white mustard (3.1) (an enzyme source) and 200 ml of water at 20°C. Stopper the flask and keep at 20°C for approximately 2 hours, shaking frequently. Add 40 ml of ethanol (3.2) and one drop of anti-foaming emulsion (3.3). Distil approximately 150 ml and collect the distillate in a 250 ml volumetric flask containing 20 ml of ammonia (3.4), making sure that the end of the condenser is immersed in the liquid. Add to the ammoniacal solution 50 ml of silver nitrate solution 0.1 N (3.5) (or more if necessary) place a small funnel over the volumetric flask and heat the mixture over a boiling water bath for 1 hour. Leave to cool, bring up to volume with water, stir and filter. Remove 100 ml of the clear filtrate, add 5 ml of nitric acid (3.7) and approximately 5 ml of ammonium ferric sulphate solution (3.8). Titrate the excess silver nitrate on return with the ammonium thiocyanate solution 0.1 N (3.6). Carry out a blank test by applying the same procedure to 2 g of finely ground white mustard omitting the sample for analysis. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Subtract the volume of silver nitrate solution 0.1 N consumed in the blank test from that consumed by the sample in solution. The value obtained gives the number of ml of silver nitrate solution 0.1 N consumed by the mustard oil in the sample. 1 ml of AgNO3 0.1 N corresponds to 4.956 mg of allyl isothiocyanate. Express the result as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
20. DETERMINATION OF BUQUINOLATE |
||||||||||||||||||||||||||||||||||||||||||||
|
(ethyl-4-hydroxy-6, 7-diisobutoxy-3-quinoline carboxylate) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of buquinolate in feeding stuffs, concentrates and pre-mixtures. The lower limit of determination is 10 mg/kg. Decoquinate interferes in the determination. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with chloroform: the extract is evaporated to dryness, the residue is dissolved in chloroform and the solution is then subjected to thin layer chromatography. The buquinolate is eluted with ethanol and determined spectrophotofluorimetrically by comparison with standard solutions. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Chloroform. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Ethanol, 96% (w/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Mixture of chloroform and ethanol: mix 10 volumes of chloroform (3.1) with one volume of ethanol (3.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Ethanol, 80% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Silica gel G for thin-layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Standard substance: pure buquinolate. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Standard solutions: |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7.1 Standard solution of 0.2 mg of buquinolate per ml: weigh out 50 mg, to within 0.1 mg, of standard substance (3.6). Dissolve in chloroform (3.1) in a 250 ml volumetric flask by warming in a water bath at 50°C. Leave to cool to room temperature, make up the volume with chloroform (3.1) and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7.2 Working standard solutions: transfer 5, 10, 15, 20 and 25 ml aliquots of the solution (3.7.1) into 25 ml volumetric flasks. Make up the volume with chloroform (3.1) and mix. Prepare immediately before use. These solutions contain respectively 0.04, 0.08, 0.12, 0.16 and 0.20 mg of buquinolate per ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flasks, with ground-glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Shaker. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Centrifuge, with tubes with ground-glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Water bath at 50°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Equipment for thin-layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Glass-plates for thin-layer chromatography, 200 x 200 mm, treated as follows: spread on the plates a uniform layer 0.5 mm thick of silica gel G (3.5) and leave to dry in the air for 15 minutes. Keep the plates in the drying oven (4.11) for 2 hours and transfer into a desiccator containing dehydrating silica gel. Ready-made plates are suitable if they give results similar to those for the plates treated as indicated above. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 0.50 ml micropipettes. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Zone collector for thin layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.9 Short-wavelength ultraviolet lamp. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.10 Spectrophotofluorimeter fitted with a xenon lamp, and two monochromators. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.11 Drying oven equipped with a fan and regulated to 100°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.12 Rotary vacuum evaporator. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the sample |
||||||||||||||||||||||||||||||||||||||||||||
|
Grind the sample so that the whole of it will pass through a sive with a 1 mm mesh (in accordance with recommendation ISO R 565). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, a quantity of the finely divided and homogeneous sample containing about 1.25 mg of buquinolate. Place the test portion in a conical flask (4.1) and add 100 ml of chloroform (3.1). Mix, stopper the flask, and shake for 1 hour using the shaker (4.2). Decant, filter and discard the first ml of the filtrate. |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer 80 ml of the clear filtrate into a beaker, or into a flask fitted to the rotary evaporator (4.12). Evaporate nearly to dryness on a water bath (4.4), dissolve the oily residue, using repeatedly a few ml of chloroform (3.1) and transfer quantitatively the liquids into a 10 ml volumetric flask, using a funnel with a thin stem. Make up the volume with chloroform (3.1) and mix. If the solution is not clear, centrifuge for 3 minutes at 3000 rpm using a stoppered tube. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Thin-layer chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Using a micropipette (4.7), deposit in spots on a plate for thin-layer chromatography (4.6), at intervals of 2 cm, volumes of 0.25 ml of the extract obtained in 5.2 and of the five working standard solutions (3.7.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
Develop the chromatogram with chloroform (3.1) until the solvent front has practically reached the upper edge of the plate, then dry with the aid of a current of air. Develop with the chloroform-ethanol mixture (3.3) until the solvent front has travelled about 12 cm. Let the solvents evaporate. Expose the chromatogram to ultraviolet light (4.9) and using a needle, mark the boundary of the strain of buquinolate spot (Rf-value 0.4--0.6). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Elution |
||||||||||||||||||||||||||||||||||||||||||||
|
Collect the silica gel from each marked zone, using a zone collector (4.8), and place in centrafuge tubes. Add to each tube 10 ml of ethanol (3.4), shake for 20 minutes, then centrifuge for 5 minutes at 3,000 rpm. Decant the clear solutions into conical flasks (4.1). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Measurement of fluorescence |
||||||||||||||||||||||||||||||||||||||||||||
|
Set the scale of the spectrophotofluorimeter (4.10) at 100 with the aid of the eluate from the most concentrated standard solution, using the excitation wavelength between 200--280 nm that gives the most intense fluorescence and an emission wavelength of 375 nm. Under these conditions, measure the fluorescence of the other eluates (5.4). From the values obtained, determine the quantity (A) of buquinolate in mg in the 10 ml of eluate from the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The buquinolate content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity in mg of buquinolate determined by spectrophotofluorimetric measurements; |
||||||||||||||||||||||||||||||||||||||||||||
|
P = weight of test portion in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
50%, relative to the higher result, for buquinolate contents between 10--20 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value for contents between 20--100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100--5,000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value for contents between 5,000--10,000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10,000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
21. DETERMINATION OF SULPHAQUINOXALINE |
||||||||||||||||||||||||||||||||||||||||||||
|
((2.4 aminobenzenesulphonamido) quinoxaline) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the content of sulphaquinoxaline in feedingstuffs, concentrates and premixtures. The lower limit of determination is 20 mg/kg. Other sulphonamides and arsanilic acid interfere with the determination. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is extracted with dimethylformamide and chloroform. The sulphaquinoxaline is partitioned into alkaline brine. After neutralisation, the amino derivative formed is diazotized and coupled with N-2-aminoethyl-1-naphthylamine. The optical density of the solution is measured at 545 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 N, N-dimethylformamide. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Chloroform. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Ethanol absolute. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Alkaline brine: dissolve 10 g sodium hydroxide and 25 g sodium chloride in water. Make up to 500 ml with water and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Hydrochloric acid, d: 1.18. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Sodium nitrite solution, 0.1% (w/v): dissolve 100 mg sodium nitrite in water, make up to 100 ml with water and mix. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Ammonium sulphamate solution, 0.05% (w/v): dissolve 500 mg ammonium sulphamate in water, make up to 100 ml with water and mix. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 N-2-aminoethyl-1-naphthylamine dihydrochloride solution, 0.1% (w/v): dissolve 100 mg of N-2-aminoethyl-1-naphthylamine dihydrochloride in 0.1% (v/v) hydrochloric acid; make up to 100 ml with the same acid and mix. Prepare immediately before use. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Standard substance: pure sulphaquinoxaline. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Standard solution: weigh out 250 mg, to within 0.1 mg, of standard substance (3.9). Dissolve in 50 ml sodium hydroxide solution (25 ml 0. 1 N sodium hydroxide solution + 25 ml water), make up to 500 ml with water and mix. Dilute 5 ml to 100 ml with water. 1 ml of this solution contains 25 µg of sulphaquinoxaline. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Conical flasks, with ground-glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Shaker. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Sintered glass funnel, porosity 3, 80 mm diameter, with filtered flask. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Separating funnels. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 50, 100, 250 and 500 ml volumetric flasks. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Test tubes approximately 150 mm x 25 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Steam bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Spectrophotometer with suitable cells (20 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the sample |
||||||||||||||||||||||||||||||||||||||||||||
|
Grind the sample so that the whole of it will pass through a sieve with 1 mm mesh (in accordance with recommendation ISO R 565). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Weight, to the nearest mg, a quantity of the finely divided and homogeneous sample containing between 0.25-1.25 mg of sulphaquinoxaline. Place the test. portion in conical flask (4.1) and add 20 ml N, N-dimethylformamide (3.1). Mix and heat the flask on the steam bath (4.7) for 20 minutes. Leave to cool under a stream of cold water. Add 60 ml of chloroform (3.2), stopper the flask and shake for 30 minutes with the aid of a shaker (4.2). |
||||||||||||||||||||||||||||||||||||||||||||
|
Filter the liquid through a sintered funnel (4.3) under milk suction. Rinse the filter flask with four 5 ml portions of chloroform (3.2) and pass the rinsings through the funnel. Transfer the filtrate to a separating funnel (4.4), rinse the filter flask with about 15 ml chloroform (3.2) and transfer the rinsings to the separating funnel. |
||||||||||||||||||||||||||||||||||||||||||||
|
Add to the separating funnel 50 ml of alkaline brine (3.4) and 5 ml ethanol (3.3). Thoroughly mix the layers avoiding emulsion formation, either by slow inversion of the funnel about 20 times or by rotating it about the horizontal axis of the stem and the stopper. Allow the layers to separate (separation is usually complete in about 15 minutes). |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer the upper layer (aqueous layer) to a 250 ml volumetric flask (4.5). Repeat the extraction of the chloroform layer with three further 50 ml portions of alkaline brine (3.4), adding each aqueous extract to the contents of the volumetric flask. Make up the volume with water and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer 25 ml of the solution to a 50 ml volumetric flask (4.5), add 5 ml hydrochloric acid (3.5), make up the volume with water and mix. Filter if necessary, discarding the first 15 ml of filtrate. Transfer 10 ml aliquots of the solution to two test tubes (4.6), A and B. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Development of colour, and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
To each tube add 2 ml of sodium nitrite solution (3.6), mix and leave to stand for 3 minutes. Add 2 ml of ammonium sulphamate solution (3.7), mix and leave to stand for 2 minutes. Add 1 ml of N-2-aminoethyl-1-naphthylamine dihydrochloride solution (3.8) to tube A and 1 ml water to tube B. Mix thoroughly the contents of each tube. By means of a water pump apply a slight vacuum to the tubes through rubber connections in order to remove dissolved nitrogen. |
||||||||||||||||||||||||||||||||||||||||||||
|
After 10 minutes measure the optical densities Ea and Eb of the solutions with the spectrophotometer (4.8) at 545 nm using water as blank. From the value Ea-Eb determine the amount (A) of sulphaquinoxaline present in the sample solution by reference to a previously prepared calibration curve (5.4). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer into a series of 100 ml volumetric flasks (4.5) volumes of 2, 4, 6, 8 and 10 ml of the standard solution (3.10) corresponding to 50, 100, 150, 200 and 250 micrograms of sulphaquinoxaline. Add 8 ml hydrochloric acid (3.5) to each flask, make up the volume with water and mix. |
||||||||||||||||||||||||||||||||||||||||||||
|
Pipette 10 ml of each solution (equivalent to 5, 10, 15,20 and 25 micrograms sulphaquinoxaline) into test tubes (4.6). Develop the colour reaction as indicated under point 5.3, first paragraph. Measure the optical densities at 545 nm using water as blank. Trace the calibration curve, using the optical density values as ordinates and the corresponding quantities of sulphaquinoxaline in micrograms as abscissae. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The sulphaquinoxaline content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity of sulphaquinoxaline in micrograms as determined by photometric measurement; |
||||||||||||||||||||||||||||||||||||||||||||
|
P = weight of test portion in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value for sulphaquinoxaline contents between 20 - 100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100 and 5000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value for contents between 5000 - 10000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
22 DETERMINATION OF FURAZOLIDONE |
||||||||||||||||||||||||||||||||||||||||||||
|
((3-(5-nitrofurfurylidenaemino)-oxazolidin-2-one)) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the quantity of furazolidone in feeding stuffs, concentrates and premixtures. The lower limit of determination is 10 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The furazolidone is extracted with acetone, after a preliminary extraction of the sample with light petroleum to remove fat. The extract is purified by chromatography on a column of aluminium oxide and the furazolidone is eluted with acetone. The acetone eluate is evaporated to dryness and the residue dissolved in pentanol. Furazolidone is then extracted from the pentanol with aqueous urea solution and the optical density of the extract is measured at 375 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Acetone |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Aluminium oxide for chromatography, neutral, 100--240 mesh, prepared as follows: stir 500 g of the aluminium oxide with 1 litre of hot water and decant the supernatant liquid. Repeat this procedure twice, and finally filter using a Buchner funnel. Dry the aluminium oxide at 105°C to constant weight. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Pentyl acetate. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Pentanol (material containing mixed isomers is acceptable). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Light petroleum, boiling range 40--60°C. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Urea solution: mix 90 g of urea with 100 ml of water, warm gently to ensure complete solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Standard substance: pure furazolidone. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Standard solution: weigh out to within 0.1 mg, 25 mg of standard substance (3.7), dissolve in acetone (3.1) in a 250 ml volumetric flask (4.1), make up to the volume with acetone (3.1) and mix. 1 ml of this solution contains 100 µg of furazolidone. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Amberglass 100 and 250 ml volumetric flasks. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Amberglass separating funnels. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Suitable extraction apparatus, e.g. Soxhlet or Twisselmann. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Extraction thimbles, approximately 25 x 80 mm or 28 x 100 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Glass tubes for chromatography, internal diameter: approximately 10 mm, length: 300 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 Steam bath. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 Spectrophotometer with suitable cells (10 mm). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
N.B. All procedures should be carried out in subdued light. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the sample |
||||||||||||||||||||||||||||||||||||||||||||
|
Grind the sample so that the whole of it will pass through a sieve with a 1 mm mesh (in accordance with recommendation ISO R 565). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, 5--20 g of the finely divided and homogeneous sample (containing not more than 1 mg of furazolidone) into an extraction thimble (4.4) and transfer it to the extraction apparatus (4.3). Extract with light petroleum (3.5), ensuring, in the case of a Soxhlet apparatus, 13--17 cycles of solvent: if other extractors are used, allow not less than 30 minutes for this stage. Remove the thimble from the apparatus, drain off the residual solvent and dry the thimble and the extracted feed in a current of warm air. |
||||||||||||||||||||||||||||||||||||||||||||
|
Place the dried thimble and contents in a clean extraction apparatus and extract with acetone (3.1), allowing at least 25 cycles of solvent when using a Soxhlet apparatus. The exact conditions for achieving complete extraction with any particular apparatus should be predetermined. Evaporate the acetone extract to a volume of 5--10 ml on the steam bath (4.6), and cool to room temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Insert a plug of glass wool into the lower end of a chromatography tube (4.5) and poke it down with a suitable rod to a thickness of 2--3 mm. Prepare a slurry of aluminium oxide (3.2) with acetone (3.1), pour into the tube and allow to settle. The prepared column should be about 200 mm in height. Allow the acetone layer to drain down to the top of the column. |
||||||||||||||||||||||||||||||||||||||||||||
|
Transfer the acetone extract obtained in 5.2 from the flask to the column, rinse the flask several times with acetone (3.1) and transfer the liquid onto the column. Place a suitable flask under the column and elute the furazolidone with acetone (3.1); the total volume of acetone used, including that used for rinsing, should be about 150 ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Extraction and measurement of the optical density |
||||||||||||||||||||||||||||||||||||||||||||
|
Evaporate the acetone eluate (5.3) just to dryness on a steam bath (4.6). (On occasions a small quantity of diacetone alcohol, produced by condensation of acetone on the aluminium oxide may be left but this will not interfere with the subsequent extractions). Dissolve the residue in 10 ml pentanol (3.4) and transfer the solution to a separating funnel (4.2). Repeat the process using 10 ml pentyl acetate (3.3) as a rinse liquid. Finally rinse the vessel which contained the extract residue with 10 ml of urea solution (3.6), add this to the separating funnel and shake fairly vigorously for 2 minutes. |
||||||||||||||||||||||||||||||||||||||||||||
|
Allow the phases to separate for a period of 3--4 minutes before transferring the aqueous extract to a 100 ml volumetric flask (4.1). Repeat the rinsing and extraction stages with four further 10 ml aliquots of urea solution (3.6) and transfer the aqueous extracts to the volumetric flask. Dilute the contents of the volumetric flask to 100 ml with urea solution (3.6) and mix. Measure the optical density of the solution in the spectrophotometer (4.7) at 375 nm against urea solution (3.6) in the reference cell. Determine the quantity of furazolidone by referring to the calibration curve (5.5). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare four chromatographic columns as described in 5.3, first paragraph. Pipette into separate columns volumes of 2.5, 5, 7.5 and 10 ml respectively of the standard solution (3.8). Wash each of the four columns with 150 ml acetone (3.1) and continue as in paragraph 5.4. Plot the calibration curve, using the optical density values as ordinates and the corresponding quantities of furazolidone in µg as abscissae. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
The furazolidone content in mg per kg of sample is given by the formula |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
A = quantity of furazolidone in micrograms as determined by photometric measurement; |
||||||||||||||||||||||||||||||||||||||||||||
|
P = weight of test portion in grams. |
||||||||||||||||||||||||||||||||||||||||||||
|
Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample must not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
50%, relative to the higher result, for furazolidone contents between 10--20 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10 mg/kg in absolute value for contents between 20--100 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
10%, relative to the higher result, for contents between 100--5000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
500 mg/kg in absolute value for contents between 100--5000 mg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
5%, relative to the higher result, for contents above 10000 mg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
23. DETERMINATION OF AFLATOXIN B1 |
||||||||||||||||||||||||||||||||||||||||||||
|
(a) ONE-DIMENSIONAL THIN LAYER CHROMATOGRAPHIC METHOD |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the level of aflatoxin B1 in the following feedingstuffs: groundnut, copra, linseed, soya, sesame, babassu palm and maize-germ oil cakes, cereals and cereal products, pea meal, potato pulp and starch. The lower limit of determination is 0.01 mg/kg (10 ppb). |
||||||||||||||||||||||||||||||||||||||||||||
|
If the presence of interfering substances hinders the determination, it is necessary to start the analysis again using method (b) (two-dimensional thin-layer chromatography). |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is subjected to extraction with chloroform. The extract is filtered, and an aliquot portion taken and purified by column chromatography on silica gel. The eluate is evaporated and the residue redissolved in a specific volume of chloroform or of a mixture of benzene and acetonitrile. An aliquot portion of this solution is subjected to thin-layer chromatography (TLC). The quantity of aflatoxin B1 is determined under UV irradiation of the chromatogram, either visually or by fluorodensitometry, by comparison with known quantities of standard aflatoxin B1. The identity of the aflatoxin B1 extracted from the feeding stuff must be confirmed by the procedure indicated. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Acetone. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Chloroform, stabilized with 0.5--1.0% of 96% ethanol (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 N-hexane. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Anhydrous diethyl ether, free from peroxides. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Mixture of benzene and acetonitrile: 98/2 (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Mixture of chloroform (3.2) and methanol (3.4): 97/3 (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Silica gel, for column chromatography, particle size 0.05--0.20 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Absorbent cotton wool, previously defatted with chloroform, or glass wool. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Sodium sulphate, anhydrous, granular. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Inert gas, e.g. nitrogen. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Hydrochloric acid solution, 1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Sulphuric acid solution, 50% (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Kieselguhr (hyflosupercel), washed in acid. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.15 Silica gel G-HR or equivalent, for TLC. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.16 Standard solution with about 0.1 µg of aflatoxin B1 per ml in chloroform (3.2) or the benzene/acetonitrile mixture (3.6), prepared and checked as indicated in Section 7. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.17 Standard solution for qualitative testing purposes containing about 0.1µg of aflatoxin B1 and B2 per ml in chloroform (3.2) or the benzene/acetonitrile mixture (3.6). These concentrations are given as a guide. They must be adjusted so as to obtain the same intensity of fluorescence for both aflatoxins. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18 Developing solvents: |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18.1 Chloroform (3.2)/acetone (3.1): 9/1 (v/v), unsaturated tank; |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18.2 Diethyl ether (3.5)/methanol (3.4)/water: 96/3/1 (v/v/v), unsaturated tank; |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18.3 Diethyl ether (3.5)/methanol (3.4)/water: 94/4.5/1.5 (v/v/v), saturated tank; |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18.4 Chloroform (3.2)/methanol (3.4) 94/6 (v/v), saturated tank; |
||||||||||||||||||||||||||||||||||||||||||||
|
3.18.5 Chloroform (3.2)/methanol (3.4): 97/3 (v/v), saturated tank. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Grinder-mixer. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Shaking apparatus or magnetic stirrer. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Fluted filter papers, Schleicher and Schull No 588 or equivalent, diameter: 24 cm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.4 Glass tube for chromatography (internal diameter: 22 mm approximately, length: 300 mm approximately), with a PTFE cock and a suitable reservoir. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.5 Rotary vacuum evaporator, with a 500 ml round-bottom flask. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.6 500 ml conical flasks, with ground-glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.7 TLC apparatus. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.8 Glass plates for TLC, 200 x 200 mm, prepared as follows (the quantities indicated are sufficient to cover five plates). Put 30 g of silica gel G-HR (3.15) into a conical flask. Add 60 ml water, stopper and shake for a minute. Spread the suspension on the plates so as to obtain a uniform layer 0.25 mm thick. Leave to dry in the air and then store in a dessicator containing silica gel. At the time of use, activate the plates by keeping them in the oven at 110°C for 1 hour. |
||||||||||||||||||||||||||||||||||||||||||||
|
Ready-to-use plates are suitable if they give results similar to those obtained with the plates prepared as indicated above. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.9 Long-wavelength (360 nm) UV lamp. The intensity of irradiation must make it possible for a spot of 1 ng of aflatoxin B1 to be still clearly distinguished on a TLC plate at a distance of 10 cm from the lamp. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.10 10 ml graduated tubes with polyethylene stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.11 UV spectrophotometer. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.12 Fluorodensitometer (optional). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Preparation of the sample (see under "Observations", Part C, point 1) |
||||||||||||||||||||||||||||||||||||||||||||
|
Grind the sample so that the whole of it will pass through a sieve with a 1 mm mesh (in accordance with recommendation ISO R 565). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Extraction |
||||||||||||||||||||||||||||||||||||||||||||
|
Put 50 g of ground mixed sample into a conical flask (4.6). Add 25 g of Kieselguhr (3.14), 25 ml of water and 250 ml of chloroform (3.2). Stopper the flask, shake or stir for 30 minutes with the apparatus (4.2) and filter through a fluted filter paper (4.3). Discard the first 10 ml of the filtrate and then collect 50 ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.3 Column clean-up |
||||||||||||||||||||||||||||||||||||||||||||
|
Insert into the lower end of a chromatography tube (4.4) a cotton or glass wool plug (3.9), fill two-thirds of the tube with chloroform (3.2) and add 5 g of sodium sulphate (3.10). |
||||||||||||||||||||||||||||||||||||||||||||
|
Check that the upper surface of the sodium sulphate layer is flat, then add 10 g of silica gel (3.8) in small portions. Stir carefully after each addition to eliminate air bubbles. Leave to stand for 15 minutes and then carefully add 15 g of sodium sulphate (3.10). Let the liquid fall until it is just above the upper surface of the sodium sulphate layer. |
||||||||||||||||||||||||||||||||||||||||||||
|
Mix the 50 ml of extract collected in 5.2 with 100 ml of n-hexane (3.3) and quantitatively transfer the mixture to the column. Let the liquid fall until it is just above the upper surface of the sodium sulphate layer. Discard this washing. Then add 100 ml of diethylether (3.5) and again allow it to fall to the upper surface of the sodium sulphate layer. Discard this washing. Then add 100 ml of diethylether (3.5) and again allow it to fall to the upper surface of the sodium sulphate layer. During these operations see that the rate of flow is 8--12 ml per minute and that the column does not run dry. Discard the liquid that comes out. Then elute with 150 ml of the chloroform/methanol mixture (3.7) and collect the whole of the eluate. |
||||||||||||||||||||||||||||||||||||||||||||
|
Evaporate the latter almost to dryness at a temperature not exceeding 50°C and under a stream of inert gas (3.11) with the rotary evaporator (4.5). Quantitatively transfer the residue, using chloroform (3.2) or the benezene-acetonitrile mixture (3.6), to a 10 ml graduated tube (4.10). Concentrate the solution under a stream of inert gas (3.11) and then adjust the volume to 2 ml with chloroform (3.2) or the benezene/acetonitrile mixture (3.6). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Thin-layer chromatography |
||||||||||||||||||||||||||||||||||||||||||||
|
Spot on a TLC plate (4.8), 2 cm from the lower edge and at intervals of 2 cm, the volumes indicated below of the standard solution and the extract: |
||||||||||||||||||||||||||||||||||||||||||||
|
--10, 15, 20, 30 and 40 µl of the standard aflatoxin B1 solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
--10µl of the extract obtained in 5.3 and, superimposed on the same point, 20 µl of the standard solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
--10 and 20 µl of the extract obtained in 5.3. |
||||||||||||||||||||||||||||||||||||||||||||
|
Develop the chromatogram in the dark with one of the developing solvents (3.18). The choice of the solvent must be made beforehand, by depositing 25 µl of the qualitative standard solution (3.17) on the plate and checking that, when developed, aflatoxin B1 and B2 are completely separated. |
||||||||||||||||||||||||||||||||||||||||||||
|
Let the solvents evaporate in the dark and then irradiate with UV light, placing the plate 10 cm from the lamp (4.9). The spots of aflatoxin B1 give a blue fluorescence. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Quantitative determinations |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine either visually or by fluorodensitometry as indicated below. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5.1 Visual measurements |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the quantity of aflatoxin B1 in the extract by matching the fluorescence intensity of the extract spots with that of one of the standard solution spots. Interpolate if necessary. The fluorescence obtained by the superimposition of the extract on the standard solution must be more intense than that of the 10 µl of extract and there must not be more than one visible spot. If the fluorescence intensity given by the 10 µl of extract is greater than that of the 40 µl of standard solution, dilute the extract 10 or 100 times with chloroform (3.2) or the benezene/acetonitrile mixture (3.6) before subjecting it again to thin-layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5.2 Measurements by fluorodensitometry |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the fluorescence intensity of the aflatoxin B1 spots with the fluorodensitometer (4.12) at an excitation wavelength of 365 nm and an emission wavelength of 443 nm. Determine the quantity of aflatoxin B1 in the extract spots by comparison of their fluorescence intensities with that of the standard aflatoxin B1 spots. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6 Confirmation of the identity of aflatoxin B1 |
||||||||||||||||||||||||||||||||||||||||||||
|
Confirm the identity of the aflatoxin B1 in the extract by the processes indicated below. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6.1 Treatment with sulphuric acid |
||||||||||||||||||||||||||||||||||||||||||||
|
Spray sulphuric acid (3.13) on to the chromatogram obtained in 5.4. The fluorescence of the aflatoxin B1 spots must turn from blue to yellow under UV irradiation. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6.2 Two-dimensional chromatography involving the formation of aflatoxin B1-hemiacetal (aflatoxin B2a) |
||||||||||||||||||||||||||||||||||||||||||||
|
NB: The operations described below must be carried out following carefully the diagram in figure 3. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6.2.1 Application of the solutions |
||||||||||||||||||||||||||||||||||||||||||||
|
Score two straight lines on the plate (4.8) parallel or two contiguous sides (6 cm in from each side) to limit migration of the solvent fronts. Spot the following solutions on the plate using capillary pipettes or microsyringes: |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point A: a volume of purified extract of the sample, obtained in 5.3, containing about 2.5 nm of aflatoxin B1; |
||||||||||||||||||||||||||||||||||||||||||||
|
--on points B and C: 25 µl of the standard solution (3.16). |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6.2.2 Development |
||||||||||||||||||||||||||||||||||||||||||||
|
Develop the chromatogram in direction I, in the dark, using the developing solvent (3.18.1) (1 cm layer in an unsaturated tank) until the solvent front reaches the solvent limit line. |
||||||||||||||||||||||||||||||||||||||||||||
|
Remove the plate from the tank and allow to dry in the dark at ambient temperature for five minutes. Then spray hydrochloric acid (3.12) along a band 2.5 cm high, covering points A and B (indicated by the hatched area in figure 3) until it darkens, protecting the rest of the plate with a glass sheet. Allow to react for 10 minutes in the dark and dry with a stream of air at ambient temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
Next, develop the chromatogram in direction II, in the dark, using the developing solvent (3.18.1) (1 cm layer in an unsaturated tank) until the solvent front reaches the solvent limit line. Remove the plate from the tank and allow to dry at ambient temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6.2.3 Interpretation of the chromatogram |
||||||||||||||||||||||||||||||||||||||||||||
|
Examine the chromatogram under UV light (4.9) and check for the following features. |
||||||||||||||||||||||||||||||||||||||||||||
|
( a ) Appearance of a blue fluorescent spot of aflatoxin B1 originating from the standard solution applied at C (migration in direction I) |
||||||||||||||||||||||||||||||||||||||||||||
|
( b ) Appearance of a blue fluorescent spot of unreacted (with the hydrochloric acid) aflatoxin B1 and a more intense blue fluorescent spot of aflatoxin B1 hemiacetal, both originating from the standard solution applied at B (migration in direction II). |
||||||||||||||||||||||||||||||||||||||||||||
|
( c ) Appearance of spots matching those described in (b), originating from the sample extract applied at A. The position of these spots is defined first by the migration distance of the aflatoxin B1 from point A in direction I (the same as that travelled by the standard applied at C), and then by the migration distances from there in direction II of the aflatoxin B1 -hemiacetal (same as those travelled by the standard applied at B). The fluorescence intensities of hemiacetal spots originating from the extract and from the standard applied at B should match. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
6.1 From the visual measurements |
||||||||||||||||||||||||||||||||||||||||||||
|
The content in micrograms of aflatoxin B1 per kg of sample (ppb) is given by the formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
X and Z are respectively the volumes in microlitres of the standard solution of aflatoxin B1 (3.16) and of the extract having a similar intensity of fluorescence; |
||||||||||||||||||||||||||||||||||||||||||||
|
S = concentration in micrograms of aflatoxin B1 per ml in the standard solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
V = final volume of the extract in microlitres, allowing for any dilution that was necessary; |
||||||||||||||||||||||||||||||||||||||||||||
|
W = weight in grams of the sample corresponding to the volume of extract subjected to column clean-up. |
||||||||||||||||||||||||||||||||||||||||||||
|
6.2 From the fluorodensitometric measurements |
||||||||||||||||||||||||||||||||||||||||||||
|
The content in micrograms of aflatoxin B1 per kg of sample is given by the formula: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
in which: |
||||||||||||||||||||||||||||||||||||||||||||
|
Y = volume in microlitres of the extract spotted on the plate (10 or 20 µl); |
||||||||||||||||||||||||||||||||||||||||||||
|
S = quantity in nanograms of aflatoxin B1 in the extract spot (proportional to the value of Y taken), deducted from the measurements; |
||||||||||||||||||||||||||||||||||||||||||||
|
V = Final volume of the extract in microlitres, allowing for any dilution that was necessary; |
||||||||||||||||||||||||||||||||||||||||||||
|
W = weight in grams of the sample corresponding to the volume of extract subjected to column clean-up. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Preparation and testing of the standard solution (3.16) |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 Determination of the concentrtion of aflatoxin B1 |
||||||||||||||||||||||||||||||||||||||||||||
|
Prepare a standard solution of aflatoxin B1 in the chloroform (3.2) or the benzene/acetonitrile mixture (3.6) with a concentration of 8 to 10 µg/ml. Determine the absorption spectrum between 330--370 nm with the aid of the spectrophotometer (4.11). |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the optical density (A) at 363 nm in the case of the chloroform solution; or at 348 nm in the case of the solution in the benezene/acetonitrile mixture. |
||||||||||||||||||||||||||||||||||||||||||||
|
Calculate the concentration in micrograms of aflatoxin B1 per ml of solution from the formula below: |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
Dilute as appropriate, away from day light, to obtain a working standard solution with a concentration of aflatoxin B1 of about 0.1 µg/ml. If kept in a refrigerator at 4°C, this solution is stable for two weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 Testing of chromatographic purity |
||||||||||||||||||||||||||||||||||||||||||||
|
Spot on a plate (4.8) 5 µl of the standard solution of aflatoxin B1 containing 8 to 10 µg/ml (7.1). Develop the chromatogram as indicated in 5.4. In UV light the chromatogram should only show one spot and no fluorescence must be perceptible in the original deposit zone. |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
The difference between the results of two parallel determinations carried out on the same sample by the same analyst should not exceed: |
||||||||||||||||||||||||||||||||||||||||||||
|
--25% related to the highest result for contents of aflatoxin B1 from 10 and up to 20 µg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
--5 µg, in absolute value, for contents greater than 20 and up to 50 µg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
--10% related to the highest value for contents above 50 µg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
9. Reproducibility |
||||||||||||||||||||||||||||||||||||||||||||
|
See under "Observations", Part C, point 2. |
||||||||||||||||||||||||||||||||||||||||||||
|
(b) TWO-DIMENSIONAL THIN-LAYER CHROMATOGRAPHIC METHOD |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the level of aflatoxin B1 in feedingstuffs not falling within the scope of method (a). The lower limit of determination is 0.01 mg/kg (10 ppb). The method is not applicable to feedingstuffs containing citrus pulp. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is subjected to extraction with chloroform. The extract is filtered, and an aliquot portion taken and purified by column chromatography on silica gel. The eluate is evaporated and the residue redissolved in a specific volume of chloroform or of a mixture of benzene and acetonitrile. An aliquot portion of this solution is subjected to two-dimensional thin-layer chromatography (TLC). The quantity of aflatoxin B1 is determined under UV irradiation of the chromatogram, either visually or by fluorodensitometry, by comparison with known quantities of standard aflatoxin B1. The identity of the aflatoxin B1 extracted from the feedingstuff must be confirmed by the procedure indicated. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Acetone. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Chloroform, stabilized with 0.5--1% of 96% ethanol (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 N-hexane. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Methanol. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 Anhydrous diethyl ether, free from peroxides. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.6 Mixture of benzene and acetonitrile: 98/2 (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.7 Mixture of chloroform (3.2) and methanol (3.4): 97/3 (v/v). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.8 Silica gel, for column chromatography, particle size 0.05--0.20 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.9 Absorbent cotton wool, previously defatted with chloroform, or glass wool. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.10 Sodium sulphate, anhydrous, granular. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.11 Inert gas, eg nitrogen. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.12 Hydrochloric acid solution, 1 N. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.13 Kieselguhr, (hyflosupercel), washed in acid. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.14 Silica gel G-HR or equivalent, for TLC. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.15 Developing solvents. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.15.1 Diethyl ether (3.5)/methanol (3.4)/water: 94/4.5/1.5 (v/v/v), saturated tank. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.15.2 Chloroform (3.2)/acetone (3.1): 9/1 (v/v), unsaturated tank. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.16 Standard solution with about 0.1µg aflatoxin B1 per ml in chloroform (3.2) or the benezene/acetonitrile mixture (3.6), prepared and checked as described in point 7 of method (a). |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
See under point 4 of method (a). |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
5.4 Two dimensional TLC |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.1 Application of the solutions (follow the diagram in figure 1). Score two straight lines on a plate (4.8) parallel to two contiguous sides (5 and 6 cm from each side respectively), to limit migration of the solvent fronts. Spot the following solutions on the plate using capillary pipettes or micro-syringes: |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point A, 20 µl of the purified sample extract obtained in 5.3; |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point B, 20 µl of the standard solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point C, 10 µl of the standard solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point D, 20 µl of the standard solution (3.16); |
||||||||||||||||||||||||||||||||||||||||||||
|
--on point E, 40 µl of the standard solution (3.16). |
||||||||||||||||||||||||||||||||||||||||||||
|
Dry in a slow stream of air or inert gas (3.11). The spots obtained must have a diameter of about 5 mm. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.2 Development (follow the diagram in figure 1). |
||||||||||||||||||||||||||||||||||||||||||||
|
Develop the chromatogram in direction I, in the dark, using the developing solvent (3.15.1) (1 cm layer in a saturated tank) until the solvent front reaches the solvent limit line. Remove the plate from the tank and allow to dry in the dark at ambient temperature for 15 minutes. |
||||||||||||||||||||||||||||||||||||||||||||
|
Then develop the chromatogram in direction II, in the dark, using the developing solvent (3.15.2) (1 cm layer in an unsaturated tank) until the solvent front reaches the solvent limit line. Remove the plate from the tank and allow to dry, in the dark, at ambient temperature. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.3 Interpretation of the chromatogram (follow the diagram in figure 2). |
||||||||||||||||||||||||||||||||||||||||||||
|
Irradiate the chromatogram with UV light by placing the plate 10 cm from the lamp (4.9). Locate the position of the blue fluorescent spots B, C, D and E of the aflatoxin B1 from the standard solution. Project two imaginary lines passing through these spots and at right angles to the development directions. The intersection P of these lines is the location in which to expect to find the aflatoxin B1 spot originating from the sample extract applied at A (figure 1). However, the actual location of the aflatoxin B1 spot may be at a point Q at the intersection of two imaginary straight lines forming an angle of about 100° between them and passing through spots B and C respectively. Determine the quantity of aflatoxin B1 in the sample extract as indicated in 5.5. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.4.4 Supplementary chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
Score two straight lines on a new plate (4.8) parallel to two contiguous sides, as indicated in the diagram in figure 1, and apply on point A (see figure 1) 20 µl of the purified sample extract obtained in 5.3 and, superimposed on it, 20 µl of the standard solution (3.16). Develop as indicated in 5.4.2. Irradiate the chromatogram with UV light (4.9) and check that: |
||||||||||||||||||||||||||||||||||||||||||||
|
--the aflatoxin B1 spots from the extract and the standard solution are superimposed. |
||||||||||||||||||||||||||||||||||||||||||||
|
--the fluorescence of this spot is more intense than that of the aflatoxin B1 spot developed at Q on the first plate. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5 Quantitative determinations |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine either visually or by fluorodensitometry as indicated below. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5.1 Visual measurements |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the quantity of aflatoxin B1 in the extract by matching the fluorescence intensity of the extract spot with that of spots C, D and E of the standard solution. Interpolate if necessary. If the fluorescence intensity given by the 20 µl of extract is greater than that of the 40 µl of standard solution, dilute the extract 10 or 100 times with chloroform (3.2) or the benzene/acetonitrile mixture (3.6) before subjecting it again to thin-layer chromatography. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.5.2 Measurements by fluorodensitometry |
||||||||||||||||||||||||||||||||||||||||||||
|
Measure the fluorescence intensity of the aflatoxin B1 spots with the fluorodensitometer (4.12), using an excitation wavelength of 365 nm and an emission wavelength of 443 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the quantity of aflatoxin B1 in the extract spot by comparison of its fluorescence intensity with that of spots C, D and E of the standard solution. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.6 Confirmation of the identity of aflatoxin B1 |
||||||||||||||||||||||||||||||||||||||||||||
|
See under point 5.6 of method (a). |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
See under point 6 of method (a). |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Repeatability |
||||||||||||||||||||||||||||||||||||||||||||
|
See under point 8 of method (a). |
||||||||||||||||||||||||||||||||||||||||||||
|
8. Reproducibility |
||||||||||||||||||||||||||||||||||||||||||||
|
See under "Observations", Part (c), point 2. |
||||||||||||||||||||||||||||||||||||||||||||
|
(c) OBSERVATIONS CONCERNING METHODS (a) AND (b) |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Defatting |
||||||||||||||||||||||||||||||||||||||||||||
|
Samples containing more than 5% fats must be defatted with light petroleum (boiling point 40--60°C) after the preparation indicated in 5.1. |
||||||||||||||||||||||||||||||||||||||||||||
|
In such cases, the analytical results must be expressed in terms of the weight of the non-defatted sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Reproducibility of the results |
||||||||||||||||||||||||||||||||||||||||||||
|
The reproducibility of the results, i.e. the variation between the results obtained by two or more laboratories on the same sample has been estimated at: |
||||||||||||||||||||||||||||||||||||||||||||
|
± 50% of the mean value for mean values of aflatoxin B1 from 10 and up to 20 µg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
± 10 µg/kg on the mean value for mean values greater than 20 and up to 50 µg/kg; |
||||||||||||||||||||||||||||||||||||||||||||
|
± 20% of the mean value for mean values above 50 µg/kg. |
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
24. DETERMINATION OF UREA |
||||||||||||||||||||||||||||||||||||||||||||
|
1. Purpose and scope |
||||||||||||||||||||||||||||||||||||||||||||
|
To determine the level of urea in feeding stuffs. |
||||||||||||||||||||||||||||||||||||||||||||
|
2. Principle |
||||||||||||||||||||||||||||||||||||||||||||
|
The sample is suspended in water with a clarifying agent. The suspension is filtered. The urea content of the filtrate is determined after the addition of 4-dimethylaminobenzaldehyde (4-DMAB) by measuring the optical density at a wavelength of 420 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
3. Reagents |
||||||||||||||||||||||||||||||||||||||||||||
|
3.1 Solution of 4-dimethylaminobenzaldehyde: dissolve 1.6 g of 4-DMAB in 100 ml of 96% ethanol and add 10 ml of hydrochloric acid (d: 1.18). This reagent keeps for a maximum period of 2 weeks. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.2 Carrez solution I: dissolve in water 21.9 g of zinc acetate Zn(CH3COO)2. 2 H2O and 3 g of glacial acetic acid. Make up to 100 ml with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.3 Carrez solution II: dissolve in water 10.6 g of potassium ferrocyanide, K4 Fe (CN)6. 3 H2O. Make up to 100 ml with water. |
||||||||||||||||||||||||||||||||||||||||||||
|
3.4 Active carbon which does not absorb urea (to be checked). |
||||||||||||||||||||||||||||||||||||||||||||
|
3.5 0.1% solution (w/v) of urea. |
||||||||||||||||||||||||||||||||||||||||||||
|
4. Apparatus |
||||||||||||||||||||||||||||||||||||||||||||
|
4.1 Shaker: approximately 35--40 rpm. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.2 Test tubes: approximately 160 x 16 mm with ground-glass stoppers. |
||||||||||||||||||||||||||||||||||||||||||||
|
4.3 Spectrophotometer. |
||||||||||||||||||||||||||||||||||||||||||||
|
5. Procedure |
||||||||||||||||||||||||||||||||||||||||||||
|
5.1 Analysis of sample |
||||||||||||||||||||||||||||||||||||||||||||
|
Weigh, to the nearest mg, approximately 2 g of the sample and place it with 1 g of active carbon (3.4) in a 500 ml volumetric flask. Add 400 ml of water and 5 ml of Carrez solutions I (3.2) and II (3.3). Shake for 30 minutes in the shaker. Make up to volume with water, mix and filter. |
||||||||||||||||||||||||||||||||||||||||||||
|
Remove 5 ml of the transparent colourless filtrate, place in test tube with a ground-glass stopper (4.2), add 5 ml of 4-DMAB solution (3.1) and mix. Place the tube in a hot-water bath at 20°C. After 15 minutes measure the optical density of the sample solution with the spectrophotometer at 420 nm (4.3). Compare with a blank test solution of the reagents. |
||||||||||||||||||||||||||||||||||||||||||||
|
5.2 Calibration curve |
||||||||||||||||||||||||||||||||||||||||||||
|
Remove volumes of 1, 2, 4, 5 and 10 ml of the urea solution (3.5), place in 100 ml volumetric flasks and make up the volume with water. Remove 5 ml from each solution, add 5 ml of 4-DMAB solution (3.1) to each of them, mix and measure the optical density as shown above in comparison with a control solution containing 5 ml of 4-DMAB and 5 ml of water free from urea. Plot the calibration curve. |
||||||||||||||||||||||||||||||||||||||||||||
|
6. Calculation of results |
||||||||||||||||||||||||||||||||||||||||||||
|
Determine the amount of urea in the sample using the calibration curve. |
||||||||||||||||||||||||||||||||||||||||||||
|
Express the result as a percentage of the sample. |
||||||||||||||||||||||||||||||||||||||||||||
|
7. Observations |
||||||||||||||||||||||||||||||||||||||||||||
|
7.1 In the case of contents of urea exceeding 3%, reduce the sample to 1 g or dilute the original solution so that there are not more than 50 mg of urea in 500 ml. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.2 In the case of low contents of urea, increase the sample as long as the filtrate remains transparent and colourless. |
||||||||||||||||||||||||||||||||||||||||||||
|
7.3 If the sample contains simple nitrogenous compounds such as amino acids, the optical density should be measured at 435 nm. |
||||||||||||||||||||||||||||||||||||||||||||
|
SCHEDULE. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
CERTIFICATE OF RESULT OF ANALYSIS. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
This is to certify that the above-mentioned sample, which was duly fastened and sealed has been analysed under the direction of the *State Chemist/*Assistant State Chemist and that the result of the analysis is as follows:-- |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
This certificate is given under the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1977. |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
*Delete as appropriate |
||||||||||||||||||||||||||||||||||||||||||||
|
GIVEN under my Official Seal, this 1st day of September, 1978. |
||||||||||||||||||||||||||||||||||||||||||||
|
JAMES GIBBONS, |
||||||||||||||||||||||||||||||||||||||||||||
|
Minister for Agriculture. |
||||||||||||||||||||||||||||||||||||||||||||
|
EXPLANATORY NOTE. |
||||||||||||||||||||||||||||||||||||||||||||
|
These Regulations, which implement certain of the provisions of Commission Directives 7½50/EEC, 71/393/EEC, 72/199/EEC, 7¾6/EEC, 74/203/EEC, 75/84/EEC and 76/372/EEC adopted pursuant to Council Directive 70/373/EEC of the 20th July, 1970, prescribe (a) the methods by which analysis of animal feeding stuffs is to be carried out for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 ( S.I. No. 302 of 1974 ) and the European Communities (Feeding Stuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1977 ( S.I. No. 246 of 1977 ) and (b) the form of certificate of the results of analyses. |
||||||||||||||||||||||||||||||||||||||||||||
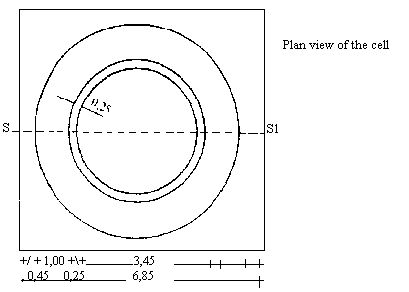
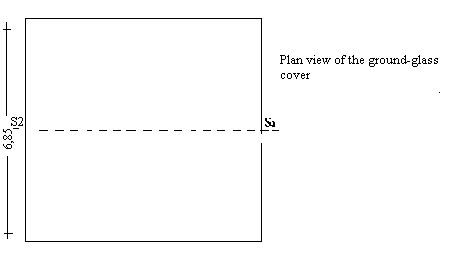
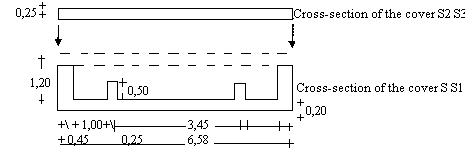
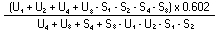
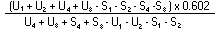
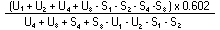
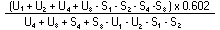
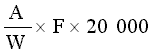
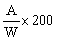
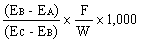
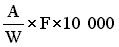
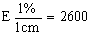
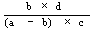
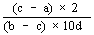
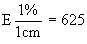
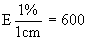
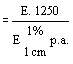
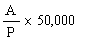
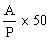
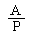
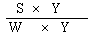
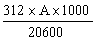 for the chloroform solution
for the chloroform solution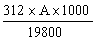 for the solution in the benezene/acetonitrile mixture.
for the solution in the benezene/acetonitrile mixture.