COVID-19: This Judgment was handed down remotely by circulation to the parties’ representatives by email. It will also be released for publication on BAILII and other websites. The date and time for hand-down is deemed to be 10am on Friday 8th October 2021
- This case concerns the formulation of a drug called sorafenib. The Defendant (‘Bayer’) is the registered proprietor of EP (UK) 2,305,255 (‘the Patent’). Claim 12 of the Patent claims the tosylate salt of sorafenib. The Claimants (‘Teva’) say that claim 12 is invalid and they seek to clear the way for their own sorafenib tosylate product.
- Originally there were a number of validity attacks pleaded but by the start of the trial, the validity attacks had reduced to just obviousness over three pieces of prior art. It is not normally necessary to address any validity attack which has been dropped and is no longer pursued. However, certain events which occurred over the weekend before the trial started had an impact on what occurred at the trial.
- One of the validity attacks pleaded by Teva was lack of novelty on the basis of (a) loss of priority and (b) intervening prior art called Lowinger. Bayer responded to that with a conditional amendment to claim 12 to claim sorafenib tosylate ‘for oral administration’. Over the weekend before trial Teva agreed to withdraw their priority attack on the basis that Bayer agreed no longer to pursue its conditional amendment to claim 12.
- Accordingly, the case was opened to me on the basis that there was no longer any issue on priority nor Lowinger and the conditional amendment had been dropped. Much of the written expert evidence was directed to attacking or defending claim 12 as proposed to be amended. During the trial, it was apparent that the battlelines as established in the written evidence were still being attacked and defended for at least some of the time, even though the target had changed. It is evident that Bayer fought its case, at least in part, on the proposed amended claim on the basis that Teva would have to prove not only that it was obvious to make sorafenib tosylate, but also that Teva had to establish that the Skilled Team would have, without invention, progressed to a formulation of sorafenib tosylate for oral administration. This might have raised an interesting issue as to what ‘for oral administration’ meant in this context, but that is not an issue I have to decide. With the proposed amendment to claim 12 having been dropped, the target was now whether it was obvious to make something which fell within claim 12.
- By the time of closing submissions, the obviousness attack was limited to a single piece of prior art called Lyons. I will come to the specifics of Lyons later, but the general picture was that sorafenib (in some form) was proving effective in Phase 1 clinical trials against three particular forms of cancer.
- In this case, the CGK is relevant to the obviousness attack and very little of it is necessary to understand the Patent. For this reason I will deal with the Patent first, then set out the CGK before moving to consider obviousness.
- The Patent is entitled ‘Aryl urea compounds in combination with other cytostatic or cytotoxic agents for treating human cancers’. It has a priority date of 3 December 2001. The field of the invention is described as follows in [0001], where the principal chemical entity is sorafenib:
[0001] This invention relates to aryl urea compounds namely the tosylate salt of N-(4-chloro-3-(trifluoromethyl)phenyl-N'-(4-(2-(N-methylcarbamoyl)-4-pyridyloxy)phenyl)urea, also in combination with cytotoxic or cytostatic agents, namely 5-fluorouracil, and their use in treating raf kinase mediated diseases such as cancer.
- Under the heading ‘Summary of the Invention’ in [0004], the Patent explains: ‘Generally, it is the overall object of the present invention to provide 5-fluorouracil in combination with sorafenib tosylate raf kinase inhibitors which will serve to…’ and then there are 8 listed benefits in the treatment of tumours.
- Although the Detailed Description starts with a general Markush formula I (A-D-B), with very wide ranges of suitable groups then being described, by [0015] the Patent states ‘The invention relates to sorafenib tosylate per se’.
- [0017]-[0019] suggest the combination(s) are effective against many forms of cancer. Then much of the description from [0020]-[0057] covers every possible form of administration of a drug, although [0026] incorporates by reference an article and pending patent application said to disclose a scalable synthesis of sorafenib tosylate.
- [0060] confirms that all the experiments reported in the Examples were performed using sorafenib tosylate, also referred to as Compound A. The experiments were conducted on female mice implanted with tumor fragments (human colon, human pancreatic, human non-small cell lung and one unidentified tumor type). Treatment was initiated once the tumors were established. The treatment in each example comprised the administration of (a) a particular cytotoxic or cytostatic agent (b) Compound A as a single agent and then (c) the concurrent therapy of both, with the percent tumour growth suppression (%TGS) measured. The picture presented by the Examples is that although Compound A on its own produced appreciable levels of TGS (e.g. 100%, 112%, 104% etc), when administered in combination, the levels of TGS were significantly better (equivalents being 229%, 222%, 133% etc). Accordingly, conclusions were drawn under each example that the anti-tumor efficacy of the concurrent therapy was either approximately additive or at least additive.
- Accordingly, the teaching in the Patent certainly established that Compound A alone had a degree of efficacy.
- Claim 1 reads as follows:
‘Use of an aryl urea compound and 5-fluorouracil or a pharmaceutically acceptable salt thereof for the preparation of a medicament for the treatment of a cancer, wherein said aryl urea compound is a tosylate salt of N-(4-chloro-3-(trifluoromethyl)phenyl-N’-(4-(2-(N-methylcarbamoyl)-4-pyridyloxy)phenyl)urea.’
- Claims 2-11 are all use claims dependent on claim 1, directly or indirectly. By way of example, Claim 3 reads:
‘The use of claim 1, wherein said cancer is colon, gastric, lung, pancreatic, ovarian, prostate, leukemia, melanoma, hepatocellular, renal, glioma, mammary or head and neck cancer.’
- Claim 12 is to:
‘Aryl urea compound, which is a tosylate salt of N-(4-chloro-3-(trifluoromethyl)phenyl-N'-(4-(2-(N-methylcarbamoyl)- 4-pyridyloxy)phenyl)urea.’
- Although it was not flagged in advance, an issue of construction of claim 12 emerged in closing submissions when I raised the point with Bayer’s counsel that the parties did not even agree on the question I had to address. Whilst Bayer naturally addressed the issue of which salts to include in a salt screen, their submissions appeared to be focussed on claim 12 as proposed to be amended - in other words Bayer was contending that to establish that unamended claim 12 was invalid Teva still needed to prove that it was obvious not just to make sorafenib tosylate but also to characterise it and then use it for oral administration to treat cancer.
- Mr Mitcheson QC for Bayer sought to justify this argument by reference to what he submitted was the invention:
15 …..The invention in this case is not about making
16 sorafenib tosylate alone. As Teva point out, the patent
17 itself does not tell you how to do that, and the experts did
18 not know how to either, the experts who were called, and the
19 patent is not directed to a medicinal chemist, it is directed
20 to a formulator. Nor does the prior art tell you how to make
21 the tosylate, nor do the CGK textbooks that everyone has
22 relied upon. That is because the technical contribution in
23 this case is not just making sorafenib tosylate, it is making
24 and then using sorafenib tosylate for oral administration to
25 treat cancer, and to obtain sufficient bioavailability to do so.
- Mr Mitcheson QC sought to draw a parallel with the approaches taken in:
i) First, Pharmacia Corp. v Merck & Co. Inc. [2002] RPC 41, where Aldous LJ at [17]-[20] rejected the argument that functional limitations should not be read into the claims. At [20] he said
‘Nobody reading the specification could believe that the ‘invention’ was the compounds claimed in claim 1. The specification makes clear that the patentees had found a class of compounds that could be made which at least had anti-inflammatory action. It was that contribution which merited a 20 year monopoly. In my view the only question capable of argument is whether the compounds in the class were chosen merely for their anti-inflammatory action or because in addition they had reduced side-effects due to them being Cox II selective.’
ii) Second, where Arnold J. (as he then was) in Idenix Pharmaceuticals Inc v Gilead Sciences Inc [2014] EWHC 3916 (Pat) at [304]-306] quoted those passages from Pharmacia but noted that the parties in that case were agreed that the validity of the claims in issue in that case should not be assessed on the basis that they were to be construed as pure compound claims but rather as claims to compounds which had particular activity.
- I note that in each of those cases, the invention was the discovery of a class of compounds in which the defining characteristic of the class was the particular activity in question, in Pharmacia, at least anti-inflammatory action; and in Idenix, anti-Flaviviridae activity.
- My task is to undertake a ‘normal construction’ of claim 12. It is unnecessary for me to set out the standard authorities but I have in mind Actavis v Lilly [2017] UKSC 48, Icescape v Ice-World [2019] FSR 5 and Liqwd Inc v L’Oreal UK Ltd [2018] EWHC 1394 (Pat), Birss J.
- Of course, each case turns on its own facts. In this case, I am entirely satisfied that unamended claim 12 does claim just the compound. I say that for the following reasons:
i) First, I remind myself that the words of these claims form part of the ‘unilateral statement by the patentee, in words of his own choosing, addressed to those likely to have a practical interest in the subject matter of his invention.’ (per Lord Diplock in Catnic).
ii) Second, in claims 1-11, the patentee had the ability to and did claim a variety of particular uses of sorafenib tosylate. These uses appear to me to cover the general statements in the specification of the ‘invention’ (save for one which I address below) in, for example, [0004], [0006] et seq., [0016]-[0017].
iii) Third, the exception is [0015] which reads ‘The invention relates to sorafenib tosylate per se.’ This is a clear signal that the patentee wishes to claim the sorafenib tosylate compound as is and without any limitation to a particular use.
iv) Fourth, consistent with that signal in [0015], claim 12 appears to be worded so as to claim sorafenib tosylate per se.
v) Fifth, the skilled person reading the Patent and the claims would understand claim 12 in effect to be a sweep-up general claim. Claims 1-11 claim the particular uses in focus in the specification i.e. to treat cancer (and particular forms) along with 5-fluorouracil or a pharmaceutically acceptable salt thereof and via various forms of administration. Having read those claims, it would be apparent to the skilled person that in claim 12 the Patentee is claiming sorafenib tosylate per se i.e. without any restriction as to use.
vi) Sixth, the skilled person would also be aware that if the Patentee had wished to formulate a claim to the invention as characterised by Mr Mitcheson QC it could easily have been done, but did not.
vii) Seventh, assuming claim 12 to be valid, I am quite sure that if a third party managed to find a use for sorafenib tosylate which fell outside claims 1-11, they would have been sued for infringement of claim 12 and one of the alleged acts of infringement would have been simply making sorafenib tosylate.
viii) Eighth, this case seems to me to be materially different to the class-type cases such as Pharmacia and Idenix, where a particular activity is the defining characteristic of the class. There is no class here, nor any need to construe claim 12 so that it is limited to a particular activity or effect. Indeed, it is readily apparent that the patentee worded claim 12 deliberately broadly so that it was not limited to any particular use, activity or effect. Furthermore, my construction of claim 12 creates no inconsistency with the description of the invention in the specification. To the contrary: as I have indicated, my construction is entirely consistent with [0015].
ix) Ninth, I asked Mr Mitcheson QC how the Court was to assess ‘sufficient bioavailability’. His answer was you have to put it [i.e. the formulated drug] into animals to ascertain it is bioavailable and also ‘you have to do enough to believe this product is worth giving to the subjects’, but these explanations were the same point as his main argument, just in a different guise.
x) Tenth, as illustrated by Bayer’s proposed amendment to claim 12, if Bayer had wanted to introduce a limitation into claim 12, it could have done so. In the end, no such limitation was before me.
xi) Overall, I find Mr Mitcheson QC’s submission to be a classic instance of Angora cat behaviour by a patentee: in this instance because only validity is in issue, the claim is presented as being narrow.
- For all these reasons, I concluded that the ‘target’ for the obviousness attack is the compound sorafenib tosylate per se.
- Professor Graham Buckton was called by Teva and Professor Henderik Frijlink by Bayer. Each of them was well qualified to assist the Court in this case, and I am grateful to both of them for their evidence and assistance. To differing degrees, each side levelled criticisms at the other’s expert, but these are best understood in the context of the arguments on obviousness, so I deal with them below.
- As appears below, the parties and the evidence concentrated very much on the CGK and characteristics of the Skilled Formulator. Whilst the Skilled Formulator is the most important member of the team, to implement the Patent the Skilled Team would have to cover a wider set of related disciplines. In particular, the Skilled Team would need medicinal chemists to make the sorafenib free base and then to make various salts of sorafenib which the Skilled Formulator selected for his or her salt screen. There was, however, no dispute over the identity of the Skilled Team. In his evidence, Professor Frijlink referred simply to the ‘formulator’, but all such references were to and should be read as referring to the Skilled Formulator.
- In this case the parties managed to agree a Primer before Experts Reports were served. At the PTR I ordered the parties to produce a statement of agreed CGK with a list of CGK issues in dispute. They did so and I am very grateful for the work done in that regard. The Statement of Agreed CGK reflected the case at the time it was prepared, addressing all the CGK relevant to claim 12 as proposed to be amended. As soon as the issue was confined to claim 12 as granted, the case centred on the preformulation stage (by which I mean the analysis leading up to the selection of the API to be formulated into a medical product) so the relevance of some parts of the CGK (relating to the formulation stage) was reduced, but I have included those parts because they help to understand some of the evidence. What follows is based on the Agreed Statement of CGK, edited to reflect some points which emerged during the trial, plus some observations of my own.
- In their reports, Professor Buckton and Professor Frijlink refer to several sources of CGK. These sources include:
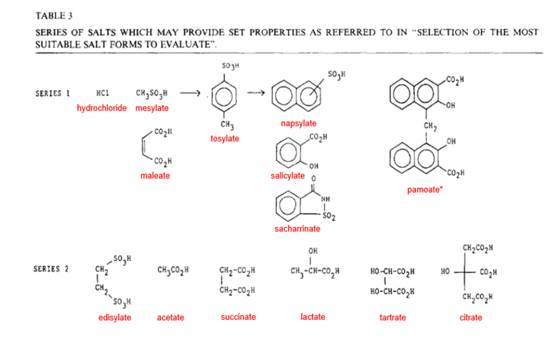
i) "Pharmaceutics: The Science of Dosage Form Design", Churchill Livingstone, by M.E. Aulton (1st Edition, 1988, 2nd Edition dated 2002 but first published on 31 October 2001, some five weeks before the priority date). This was described by Professor Buckton as a well-known textbook which outlines the basic principles of formulation. In his oral evidence he referred to it as a basic undergraduate textbook. Professor Frijlink said it was widely recognised as one of the leading textbooks on formulation science. Whilst I accept that some formulators in real-life would have ordered and obtained a copy of the second edition by the priority date, I think it is more likely and I find that the majority would have continued to work from and consult the first edition. It is unrealistic, in my view, to suggest that formulators keep their working libraries under constant review. One difference between the relevant chapters of the first and second editions was in the table of possible pharmaceutical salts. In the second edition, table 8.4 was shorter than table 13.4 in the first edition, with the result that the tosylate counterion did not feature (although it was referred to in the text of both editions - see below).
ii) “Salt Forms of Drugs and Absorption” by Bighley et al from Encyclopedia of Pharmaceutical Technology (edited by J. Swarbrick and J.C. Boylan), Vol. 13, 1996, 453-499 (“Bighley”). This is a more specialised text, but one which both experts were familiar with prior to their involvement in this case. Professor Frijlink described it as an authoritative overview of drug development, dosage, manufacturing and regulation.
iii) “Pharmaceutical salts” by S.M. Berge, L.D. Bighley and DC Monkhouse, from Journal of Pharmaceutical Sciences, American Pharmaceutical Association, January 1977, Volume 66, No. 1 (“Berge”). Berge seems to have been an early attempt to identify some general trends in how to successfully achieve a desired combination of properties in salt formation, a process they describe as a difficult semiempirical choice. Berge contains an analysis of which salts featured in drugs approved by the FDA down to 1974, a table which is reproduced in Gould.
iv) “Salt selection and optimisation procedures for pharmaceutical new entities” by R.J. Bastin et al, Organic Process Research and Development, 2000, 4, 427-435 (“Bastin”). This is an article from authors at Aventis Pharma which Professor Frijlink identified when asked by the Defendant’s solicitors about resources relating to salt selection in late 2001, also referred to by Professor Buckton. Bastin contains an overview of salt selection, followed by analysis of comparative results of salts of three different drugs and then tests for the preclinical phase. As Professor Frijlink pointed out, at least part of the reason for the article was to draw attention to an early high-throughput screening method apparently developed at Aventis Pharma.
v) “Salt selection for basic drugs” by Gould, International Journal of Pharmaceutics, 33 (1986) 201-217 (“Gould”) (GB-07). A journal article from Gould at Pfizer, which Professor Buckton was familiar with prior to his involvement in this case, as was Professor Frijlink, although he did not refer to it in his first report and agreed, in his second report, that Gould could be added to the list of CGK sources, albeit (like Berge) it was an older resource by the Priority Date. Gould identifies a number of pivotal issues for salt selection - melting point, drug solubility, salt hydrophobicity - which he suggests need to be considered before deciding on the most suitable range of salt forms to prepare, whilst emphasising the need to consider ‘balance’. He then clusters the conjugate acids into groups for addressing specific issues. He concludes by saying ‘The balance required in assessing the correct salt form to progress into drug development makes it a difficult semi-empirical exercise.’ To assist in salt selection he provides an appendix providing details of ‘a wide series of conjugate acids, including details of structure, melting point, pKa, LD50 and examples of use…’
- The experts were agreed that none of these resources form part of the CGK in their entirety, but generally provide guidance and reflect principles which would have been known by the Skilled Formulator at the Priority Date. Due to the focus placed on some of these sources in the expert evidence, I will need to return to consider more precisely what content was CGK.
- An early stage in the pharmaceutical development of a new chemical entity (“NCE”) will be to characterise the NCE in its native form (i.e. the form in which it is received from the medicinal chemists; for ionisable compounds this is the free acid or free base form), in terms of physicochemical properties. This analysis phase is part of what is commonly referred to as preformulation. The substance which is the active ingredient in the particular dosage form is referred to as the active pharmaceutical ingredient (“API”). This may or may not be a salt form of the NCE.
- Preformulation tests on the NCE often include:
i) the study of solubility (testing in aqueous and non-aqueous solvents and at different pH). Analysing solubility is considered to be one of the most important aspects of preformulation;
ii) the study of chemical stability (testing in solution (at different pH) and in the solid state (at different temperatures and humidity levels) and the effect of both oxygen and light); and
iii) other tests relating to physical properties of the compound, such as pKa (the strength of an acidic or basic group), hygroscopicity, and melting point and an assessment of whether a compound is crystalline.
- An assessment of permeability (partitioning between water and octanol, or use of cell culture models for determining absorption) will often also be undertaken on the NCE.
- The Skilled Formulator having analysed the NCE in its “as received” form might identify key issues which present a concern from a formulation perspective, for example whether the aqueous solubility is very low or the NCE is unstable or will not crystallise, and find ways to try to improve them.
- For oral dosage forms, as discussed further below, it is important that the API has some aqueous solubility at the pH of fluids within the gastrointestinal tract and is also sufficiently stable to be formulated. On the assumption that the NCE is an acid or a base, which it usually will be, then a common approach is to address potential concerns relating to solubility and stability by salt screening and selection. The reasons for salt usage are because this is the most straightforward way in which the Skilled Formulator can alter the solubility, stability and processability of the NCE.
- It is the role of the Skilled Formulator to ensure that the API is formulated into a medicinal product that is effective, safe, stable and ensures ease of administration. This means using a combination of the API with inactive substances (referred to as excipients). There would be an early estimate of dose. However, the Skilled Formulator will be mindful that the dose of the API is likely to change based on the results of early clinical trials, and they would therefore use formulation strategies that are best able to accommodate dose changes. Typically, but not exclusively, processes such as wet granulation and tabletting are amenable to variation in dosage.
- The principal objective of the formulator is to take the candidate compound and create a dosage form which is stable and can deliver a suitable and reproducible amount of the active ingredient into the blood stream of the patient.
- Routes of drug administration include oral, parenteral, inhaled and topical administration. The oral route, most usually by use of a solid oral dosage form (e.g. tablets and capsules), is the most frequently used route of drug administration. It is considered the simplest, most convenient and safest way of API administration, though it requires at least some of the administered API to be capable of passing through the gastrointestinal tract, absorptive membrane and first passage of the liver without being inactivated.
- Use of oral dosage forms requires consideration of the absorption process from the gastrointestinal tract (discussed further below).
- Oral dosage forms include tablets, capsules, suspension, solutions and emulsions.
- Clinical considerations with respect to dosage form selection involve ease of administration, compliance issues and the type of disease being treated. For cancer patients who were well enough to take their own medication, oral dosage forms would have been preferred.
- The human gastrointestinal tract involves a number of different structures with different physiological features. Whilst there can be variability in the conditions through the gastrointestinal tract, typically the pH in the stomach is around pH 1 - 3.5, in the small intestine around pH 5 - 7 and in the large intestine around pH 6 - 7.5.
- For an orally administered dosage form to be efficacious the API must be released and absorbed into the bloodstream after administration. This takes place in the gastrointestinal tract, where the API must (a) dissolve into the gastrointestinal fluids and (b) pass through the membrane (predominantly the intestinal membrane which is the region best suited for absorption) to enter the bloodstream.
- The amount of API that reaches the blood plasma relative to the amount administered is known as its bioavailability. The extent of bioavailability from an oral dosage form will be a consequence of the physicochemical properties of the API, the formulation and the method of manufacture.
- Different routes of delivery (i.e. other than oral) can also result in different bioavailability.
- For orally administered drugs, what is generally seen as important is not achieving as close to 100% bioavailability as possible but producing a formulation with a level of bioavailability such that enough API enters the systemic circulation so as to be safe, effective and most importantly, reproducible. Consistency in bioavailability is most important as variability can result in different therapeutic outcomes and side effect severity.
- The solubility and the permeability of an API are key factors in determining its bioavailability. The two most significant processes that affect bioavailability of an orally administered API are the dissolution of the API into solution (which is not exclusively, but to a large extent, influenced by the solubility of the API) and the absorption of the API into the plasma (which is due to the permeability of the API through the semipermeable membranes of the gastrointestinal tract to the systemic circulation).
- Permeability is generally an inherent property of the API that can only be fundamentally altered by changing the molecular structure of the API, rather than e.g. by forming a salt.
- Solubility is one of the key properties of a candidate compound that the formulator will be concerned with.
- Solubility can be defined as the amount of a substance that will dissolve in a given volume of solvent at a specified temperature when at equilibrium. The material that dissolves in the solvent is referred to as the solute. Solubility measurements can be given by reference to the mass of an API that may dissolve in a given volume of solvent (e.g. milligrams per millilitre). The greater the mass of API that may dissolve in a given volume of solvent, the better its solubility.
- All systemically active APIs (i.e. APIs which are present throughout the body, typically via the circulatory system, rather than being locally administered) must exhibit at least limited aqueous solubility for absorption in the body and therefore therapeutic efficacy, regardless of their route of administration.
- Solubility often varies at different temperatures and pH values. It may therefore be measured at different temperatures, including at room temperature and at body temperature. If oral administration is being considered, solubility should also be measured at different pHs to reflect the pH at different places in the gastrointestinal tract.
- The API present in solid oral dosage forms such as tablets or powder capsules needs to dissolve into the gastrointestinal fluids before it can be absorbed across the gastrointestinal membrane. The process by which an API dissolves from a dosage form is called dissolution. The API must first come into contact with the solvent, dissolve into it and then move away from the remaining solid by diffusion. As dissolved molecules of API diffuse out of the diffusion layer which surrounds the dissolving API, further dissolution from the remaining solid will occur.
- In the case of a solid oral dosage form such as a tablet, dissolution will usually be preceded by disintegration to release smaller particles into the gastrointestinal fluids (these particles may include other excipients, not just API).
- Solubility is a key determinant of dissolution rate. Further factors that can affect the dissolution rate of an API can include the particle size (which affects the surface area of the API available to dissolve), the wettability (how well a solid surface and a liquid maintain contact, which affects surface area), the form of an API (e.g. salt or free form, crystalline or amorphous, which affect solubility) and the molecular weight of the API.
- Different salts have different equilibrium solubility at certain pH values. Amorphous forms are the same chemical substance as their crystalline form, so they do not have different equilibrium solubility, although an amorphous form may dissolve faster than its crystalline form and may (transiently) yield a supersaturated solution, thus appearing to have a different solubility.
- The pH of the solution can have a significant impact on the solubility of an API. The solubility of weakly acidic APIs increases with increasing pH, so as the API moves down the gastrointestinal tract from the stomach to the intestine its solubility will increase. Conversely, the solubility of weak bases decreases with increasing pH, so as the API moves down the gastrointestinal tract its solubility will decrease. Depending on the pKa, the rate of dissolution for poorly soluble weak bases is therefore likely to be considerably higher in the stomach than in the small intestine, and vice versa for weak acids.
- Dissolution rate is directly proportional to the available surface area and solubility of the API. As can be seen from the Noyes-Whitney equation set out below, the factors which affect dissolution rate are available surface area for dissolution (A), solubility of the API in the diffusion layer (Cs), concentration of dissolved API at a particular point in time in the bulk solution (Ct) and a factor (k) relating to diffusion of the dissolved material away from the dissolving dosage form and the thickness of the unstirred layer.
Dissolution rate = Ak(Cs-Ct)
- The BCS provides a system by which pharmaceutical compounds may be categorised based on their solubility and permeability in the gastrointestinal tract. It derives from a paper by Amidon et al from 1995. Although the BCS was CGK, the experts were agreed that the entire content of Amidon’s paper would not be. The BCS allocates pharmaceutical compounds to one of four "classes" in accordance with these properties:
|
 Class Class |
Solubility |
Permeability |
|
I |
High |
High |
|
II |
Low |
High |
|
III |
High |
Low |
|
IV |
Low |
Low |
- An API is considered to be highly soluble where the highest dose strength is soluble in 250mL or less of aqueous media over the pH range 1-8. If the volume of aqueous media required to dissolve the API in pH range 1-8 is greater than 250 mL then the API is considered to have low solubility.
- At the Priority Date, the majority of NCEs that entered development were BCS Class II, i.e. had low solubility and high permeability. Formulating NCEs with this combination of characteristics was therefore something that the Skilled Formulator would be used to doing on a regular basis.
- For Class II, as a result of poor solubility, drug dissolution will usually be the rate-limiting step for absorption.
- If drug compounds are in Classes II or IV then the Skilled Formulator will want to consider technical options to improve the less advantageous properties of the drug.
- A commonly used way (if not the most common way) to improve the aqueous solubility of a poorly soluble API is by preparation of a salt. Often a major improvement in solubility at some pH values can be achieved, compared to the free acid or base, by forming a suitable salt.
- The majority of new APIs approved by the regulatory authorities and marketed as medicinal products were in a salt form at the Priority Date (and still are today).
- The process of salt formation involves pairing the NCE with an appropriate counterion (i.e. by reacting an acidic NCE with a base or by reacting a basic NCE with an acid). Other factors relevant when selecting salts for a salt screen include the safety of the counterion and the route of administration.
- If the aim of salt formation is to improve solubility, then the Skilled Formulator will ideally want to ensure that solubility is improved at pH values within the pH range experienced by the API as it travels down the gastrointestinal tract (i.e. within the range of pH 1 to 6.8) and at body temperature.
- All ionisable NCEs have what is referred to as a “pH solubility profile”. Solubility is at a minimum when the substance is unionised and increases as the compound becomes ionised in solution.
- Solubility reaches a maximum when essentially fully ionised. Weak acids are unionised at low pH and become ionised at higher pH. Conversely, weak bases are unionised at high pH and are more soluble at low pH.
- Acid addition salts of any basic compound alter the solubility below the pKa of that compound to different extents.
- Although addressing poor aqueous solubility is the most common reason to conduct a salt screen, salts are also prepared to address other physicochemical and biological concerns, including instability, and processability of the API.
- The process of selecting counterions to try and pair with a free acid or base is often called salt selection. The process of attempting to make salt forms using selected counterions and investigating the properties of these salts is called a salt screen. Typically, a formulator would consider the properties of the free acid or base drug compound under consideration (as discussed above) and select salts they consider would be a good match to include in the salt screen. Some pharmaceutical companies may also have had salts they routinely used more or less frequently.
- To form a salt, which is a solid state material, the NCE and counterion are dissolved together and brought out of solution as the salt. The salts with the most desirable properties, which if the issue being addressed is low solubility would include solubility enhancement, are selected for further investigation. The number of salts is reduced by subsequent testing.
- The essential prerequisite for salt formation is the presence of ionisable (i.e. acidic or basic) functional groups in the API’s structure that allow sufficient ionic interaction between the API and the counterion.
- For successful formation of a salt to be possible the pKa of an acidic counterion must be lower than the pKa of the ionisable group of a basic API. Conversely, the pKa of a basic counterion must be higher than the pKa of the ionisable group of an acidic API. Accordingly, a formulator will consider the pKa of the acidic/basic group on the API and that of the potential counterion.
- This means that for some basic APIs, i.e. those which have a low pKa, there is a more limited number of pharmaceutically acceptable acidic counterions that can be used for salt formation than for other APIs with a higher pKa (i.e. a stronger base).
- For weak bases with a pKa of less than about 3, the salts formed can sometimes have poor physical stability in the solid state as they may easily disproportionate back to the free form with atmospheric moisture in the solid state, or when in contact with excipients in a drug product.
- However, even for NCEs with such low pKa values, salt formation would be the first choice for improving solubility.
- It is possible to predict pKa using theoretical calculations or to measure it with standard experimental procedures. The formulator would be aware that the pKa values arising from such calculations might lead to different values.
- Different counterions would generally be screened for salt selection simultaneously in order to save time overall (doing each test sequentially would take too long in the context of commercial development). There is no absolute number of acids that would be selected for a salt screen, but formulators in industry would make a reasonable number of salts in parallel.
- Pharmaceutically acceptable acids/bases are identified in the texts listed at paragraph 26 above. When looking to formulate a weak base, the screen would include a selection of acid counterions of suitable pKa from the acids described in these texts. These texts also give overall guidance on the expected properties of salts when they are made. The guidance shows that some acids will be expected to make a salt less soluble, and others will be expected to enhance solubility of a poorly soluble basic compound.
- Whilst the expectations and general trends exist, numerical values (for example, for solubility) cannot be obtained prior to the salt being formed and it would not necessarily be possible for the Skilled Formulator to predict which particular salt would have the optimum properties to use in a particular pharmaceutical dosage form. Salt selection is therefore a matter of testing of salts in order to obtain salts with the optimal balance of properties. The selection of acids with which to make salts of an API will be guided by the concepts in the references noted above, but the salts will need to be made and tested to ascertain which one(s) will have the best overall properties.
- By 2001, the hydrochloride would almost invariably be included in a salt screen for a basic drug. However, a common ion effect can be observed with hydrochloride salts meaning that they do not necessarily enhance the solubility of poorly soluble drugs in the stomach.
- The common ion effect is where the solubility of one material is limited by an ion which is already in solution. For example, the stomach is rich in hydrochloric acid which may limit solubility of some hydrochloride salts.
- One of the tests which would routinely be carried out during formulation development (although not typically as part of an initial salt screen) is a dissolution rate test. Dissolution rate tests require a greater quantity of drug substance than solubility tests and so typically came at a slightly later stage of development.
- If further improvement in dissolution rate is required, the Skilled Formulator would consider other techniques, as discussed below.
- Reducing the particle size of the API into the region of 1-10 microns.
- This includes the use of excipients such as disintegrants and surfactants.
- Nanosizing reduces particle sizes to sub-micron levels. Nanosizing had significant disadvantages in terms of the availability and cost of the specialised equipment, and the process could cause manufacturing difficulties.
- The liquid filled capsule will contain the drug compound dissolved or dispersed in an oily liquid medium containing surfactants.
- Liquid filled capsules can lead to significant increases in the absorption speed of a drug, but they are technically more complex and more expensive than tablets to manufacture so are commercially a less appealing option.
- A poorly soluble drug could also be formulated as a solid dispersion.
- Solid dispersions can increase the apparent solubility by having a rapid dissolution rate. This can yield a higher amount of absorption of the drug, compared to crystalline forms, but again is a more complex and expensive technology.
- The use of cyclodextrins to increase the aqueous solubility and absorption of a poorly soluble drug could also be considered by the formulator.
- Cyclodextrins are relatively large molecules made up of multiple glucose units in a torus-like cyclic arrangement.
- The parties identified two CGK points as being in dispute - dose number and company standard operating procedures - which I deal with below. However, I also introduce in this section a further topic - the selection of salts to include in a salt screen - on which it became apparent that each side contended the CGK went further than the Agreed Statement in various ways.
- The parties were at odds as to whether familiarity with the concept of the dose number would form part of the skilled formulator’s CGK or not. The Claimants’ position was that it would not, the Defendant contended it would. Although Professor Frijlink made some strong assertions based on dose number, the concept seemed to relate to the formulation stage, now much less relevant. Indeed, it was Professor Buckton’s evidence that information as to the dose number is only available to formulators of generic medicines with knowledge of the innovator product to hand, hence, he said, it was not relevant to the salt selection process for NCEs. I am inclined to agree. Furthermore, I do not see that this dispute matters to anything I have to decide.
- The Defendant’s position was that formulators in industry would have had access to company standard operating procedures or internal manuals (together ‘CSOPs’) relating to pre-formulation and formulation and that these would have broadly reflected the CGK.
- The Claimants’ position was that to the extent that standard operating procedures or internal manuals relating to pre-formulation and formulation existed, there is no evidence in the case to suggest their specific content and it therefore cannot be accepted that any or all such manuals would have broadly reflected the CGK.
- This dispute appears to me to go nowhere. On the Defendant’s case, if CSOPs broadly reflected the CGK, they add nothing. Furthermore, there were no CSOPs in evidence. This point, however, may have been raised in an attempt to bolster some of Professor Frijlink’s evidence about what was done during his time at Solvay. I will deal with those pieces of evidence as they arise below.
- Although this topic was not identified by the parties as being an issue of CGK in dispute, it became apparent to me that the sections above (reproduced from the statement of agreed CGK) concerning the selection of salts to include in a salt screen are incomplete. From the agreed base, each of the experts went further in different ways. Certain disputes between the experts emerged more clearly in the course of cross-examination, and most concerned the balance which the Skilled Team or Formulator would have to strike between competing considerations when considering which salts to include in their salt screen.
- Both experts in their reports, albeit to slightly differing degrees, delved into particular passages in the documents listed at paragraph 26 above. In my view, this was particularly the case in the reply report from Professor Frijlink. The searching for and reliance on particular passages or sentences from those texts is entirely understandable for an expert witness wishing to locate material to support a particular opinion. Understandable also, in the circumstances of this litigation, was a focus on the tosylate salt and whether it would or would not be included in a salt screen.
- I remind myself however that the Skilled Team, when selecting the salts to include in their salt screen, would not have focussed on tosylate in particular, either to include it or to exclude it, and such focus carries a very distinct risk of introducing illegitimate hindsight into the analysis. Furthermore, the Skilled Team would not have reached or even sought to reach any sort of definitive view on the suitability of potential candidate salts, precisely because of the considerations mentioned in paragraph 79 above. This general point underpins my approach to certain parts of the evidence, which I address below.
- This is familiar territory, but it is nonetheless useful for me to remind myself of the applicable principles which I can take from the Judgment of Arnold J (as he then was) in Allergan Inc. and anor v. Aspire Pharma Ltd [2019] EWHC 1085 (Pat) where he described "the overall tenor" of the Supreme Court's judgment as "confirm[ing] the approach which had previously been adopted by the courts to this question". Arnold J. went onto to distil five points from that Judgment, at his [97]-[102]:
97. First, at [60] and [93]-[96] Lord Hodge endorsed, while not mandating, the use of the structured approach set out in Windsurfing International Inc v Tabur Marine (Great Britain) Ltd [1985] RPC 59 as reformulated in Pozzoli SPA v BDMO SA [2007] EWCA Civ 588, [2007] FSR 37 at [33].
98. Secondly, at [63] Lord Hodge endorsed, while emphasising that it was not exhaustive, the statement of Kitchin J (as he then was) in Generics (UK) Ltd v H Lundbeck A/S [2007] EWHC 1040 (Pat), [2007] RPC 32 at [72]:
"The question of obviousness must be considered on the facts of each case. The court must consider the weight to be attached to any particular factor in the light of all the relevant circumstances. These may include such matters as the motive to find a solution to the problem the patent addresses, the number and extent of the possible avenues of research, the effort involved in pursuing them and the expectation of success."
99. Thirdly, at [65] Lord Hodge agreed that it was relevant to consider whether something was "obvious to try", saying that "[i]n many cases the consideration that there is a likelihood of success which is sufficient to warrant an actual trial is an important pointer to obviousness". He nevertheless endorsed the observation of Birss J at first instance that "some experiments which are undertaken without any particular expectation as to result are obvious".
100. Fourthly, at [69] Lord Hodge said that "the existence of alternative or multiple paths of research will often be an indicator that the invention ... was not obvious", but nevertheless endorsed the statement of Laddie J in Brugger v Medic-Aid Ltd (No 2) [1996] RPC 645 at 661: "[I]f a particular route is an obvious one to take or try, it is not rendered any less obvious from a technical point of view merely because there are a number, and perhaps a large number, of other obvious routes as well."
101. Although Lord Hodge did not explicitly make the point, it is implicit in his endorsement of this statement that it remains the law that what matters is whether the claimed invention is obvious from a technical point of view, not whether it would be commercially obvious to implement it.
102. Fifthly, at [70] Lord Hodge confirmed that the motive of the skilled person was a relevant consideration. As he put it:
"The notional skilled person is not assumed to undertake technical trials for the sake of doing so but rather because he or she has some end in mind. It is not sufficient that a skilled person could undertake a particular trial; one may wish to ask whether in the circumstances he or she would be motivated to do so. The absence of a motive to take the allegedly inventive step makes an argument of obviousness more difficult."
- Understandably in the context of this case, both sides placed particular emphasis on the ‘could/would’ distinction, the subject of Lord Hodge’s fifth point.
- For its part, Bayer submitted:
‘…it is not enough that a skilled person could make a particular choice, to render that choice obvious. The question is whether there is something that will in fact spur him/her on to do it - whether, in all the circumstances, s/he would do it.’
- Teva draw attention to the following passage in the judgment of Birss J (as he then was) in Hospira v Genentech [2014] EWHC 3857 (Pat) at [227]-[234]:
“227. Hospira submitted, based on Prof Halbert's evidence, that the claimed formulation was one of the formulations that the skilled person could have derived by routine means. Genentech emphasised by reference to case law in the EPO (citing the Case Law of the EPO (7th Ed 2013 at I.D.5)) and cases in this jurisdiction that the question is whether the skilled person would have arrived at the claimed invention, not whether they could have. Genentech argued that Hospira's submission was the highest that Hospira's case could be put and since it was put on the basis of the word "could", that was not enough to establish a lack of inventive step. This argument raises a number of points.
228. First, while the submission is an accurate way of stating part of Hospira's case, it is not the whole of the obviousness case. That case includes other elements, in particular the precise status of trehalose, histidine and polysorbate 20 in the common general knowledge of the skilled team.
229. Second, the law of obviousness cannot be accurately summarised simply by stating that the question is whether the skilled person would have arrived at the claimed invention, not whether they could have. The issue is multifactorial and based closely on the particular circumstances.
230. Third, the word "would" is not always straightforward. Sometimes asking simply if a skilled person "would" do something risks placing too much weight on what are really minor or irrelevant factors like cost, instead of focussing on the technical issues. Moreover, the well-known 9 ½ inch plate is not something a skilled person would make. It is more accurate to say that it is not patentable because the skilled person could make it without any inventive step.
231. In other cases the difference between could and would is important. If the outcome rides on the result of a single experiment, the fact the skilled person could carry it out does not usually mean the invention is obvious. One often needs to ask if they would carry out the test in the expectation of a positive result.
232. This dependence on the facts is the reason why the passage from Kitchin J's judgment in Generics v Lundbeck, approved in the House of Lords in Angiotech, is significant and why the Court of Appeal in Medimmune emphasised that there is a single statutory test, repeating at paragraph 95 Lord Walker's concern (in HGS v Lilly) about the utility of elaborate judicial exposition.
233. Fourth, real skilled teams faced with trying to formulate lyophilised trastuzumab would do many different things. They would have their own personal experience and idiosyncrasies and their own resource limitations. I am quite sure if one compared a number of real skilled teams side by side, they would test different combinations of excipients in a first and second screen. Some teams who found unpromising results in the first and second tier screen would continue past a second tier screen, others might not. Some real teams might never test polysorbate 20 or histidine at all. For all we know polysorbate 80 is just as good as polysorbate 20. Thus a real team which started with polysorbate 80 might never see a need to test another surfactant. Equally for all we know polysorbate 80 does not work with trastuzumab (in which case I am quite sure nearly every team would at least try polysorbate 20 when they encountered such problems). The only evidence before the court about what works and what does not is the data in the patent. Given the empirical nature of this field, the outcome of experiments which have not been carried out cannot be predicted.
234. My conclusions on the could/would argument are as follows. It is not true to say that a real team would arrive at a formulation consisting of polysorbate 20, histidine and trehalose. It would be idle to pretend otherwise and Hospira do not do so. But what Hospira's submission is getting at is that the claimed result can be reached by the application of nothing other than routine approaches applied to excipients which were part of their common general knowledge. In my judgment on the facts of this case that is correct.”
- This analysis was upheld on appeal by Floyd LJ at [2016] EWCA Civ 780 at [50]-[52].
“50. Next, I must deal with the could/would debate. I have already explained why I do not accept that it is necessary in every case for the court to conclude that the skilled person acting only on the basis of the prior art and his common general knowledge would arrive without invention at the precise combination claimed. Given that the screening methods were part of the common general knowledge, that the tests involved were routine, that the excipients were common general knowledge excipients and that there was no a priori reason why a successful lyophilised formulation could not be made, it seems to me that it was beyond argument that the claimed combination in this case was one that could be made by the skilled team. The question is whether this is the type of case where it is necessary to go further and ask whether the skilled person would necessarily have made the precise combination claimed.
51. In an empirical field it will be seldom be possible to predict in advance that any individual experiment will work. In many cases, the fact that a routine screening exercise could be carried out will be inadequate to establish obviousness. Nevertheless, on the facts of an individual case such as the present, the team may have a reasonable degree of confidence that a series of experiments will produce some which will work. To impose a requirement that the skilled team must be able to predict in advance which would be the successful combinations is wholly unrealistic. It would lead to the grant of patents for a whole variety of combinations which in fact involved no inventive effort.
52. It was inherent in Mr Tappin's argument that it is only in the case of an invention which is an arbitrary selection that it is open to the court to find obviousness on the basis of a "could" test. As the judge had held that the combination here was not arbitrary, there could be no finding of obviousness. I do not agree. The case where the invention is simply a choice of one candidate from a field where all the individual candidates are the same is an extreme case. The present case is one where the skilled person would expect there to be a range of different results, some good and some bad, but that there was no invention in embarking on a screening process to pick out the good from the bad. The fact that one could not say in advance which the good ones would be does not, in every case, foreclose a finding of obviousness.”
- Bayer accused Teva of relying on Hospira v Genentech and Ranbaxy to try to distance itself from what Bayer called the ‘standard could/would approach to obviousness’. Bayer also submitted that those cases cannot ‘trump’ either the Supreme Court’s decision in ICOS nor the consistent approach of the EPO’s TBA which distinguishes what a skilled person merely could do from the obvious things s/he would do. Finally on this point, Bayer submitted that neither Ranbaxy nor Hospira is analogous on the facts.
- I do not see any conflict between ICOS and the approaches taken by the Courts in either Ranbaxy or Hospira, or in the EPO TBAs for that matter. Each case turns on its own facts. However, the arguments related by Birss J. and Floyd LJ. in Hospira, for example, reflect some of the points made in this case. Whether those arguments succeed in this case depends on the circumstances and evidence presented in this case.
- I realise that I must proceed on the basis of the evidence led before me in this case. Nonetheless, I set out those passages because of their particular relevance in terms of the approach and principles.
- Bayer sought to make a particular point derived from Lord Hodge’s third, fourth and fifth points. It submitted that in a case such as the present, where part of the dispute is about what options the skilled person would select as candidates to test, and what s/he would then choose to proceed with in light of those tests, it is clear that such a test would proceed first with those options the skilled person would consider hopeful ones. Bayer submitted that where there is a good technical reason to think a particular option is not likely to be a fruitful one, that option gets relegated further down (or even off) the notional list.
- This submission is fine so far as it goes, but it is important that the analysis is not infected by hindsight. Furthermore, this submission had more relevance to the arguments over claim 12 as proposed to be amended.
- As in most, if not all, cases of obviousness, the patentee here (Bayer) majored on hindsight. Indeed, in its final submissions, Bayer identified no less than 20 steps which it said the uninventive Skilled Team would have to undertake to get to the target (but it is important to note that the ‘target’ which Bayer had in mind was claim 12 as proposed to be amended). Naturally whilst Bayer accepted that certain of these steps might have been obvious, it contended that others were not and overall it characterised the process as a classic Technograph step-by-step approach. Furthermore, Bayer warned me, one has to guard against deconstructing the invention into a series of small steps, each of which is then portrayed as a small iteration.
- It is easy to raise the Technograph point against any work which necessarily requires a number of steps to be taken. In every case, the question is whether the combination of steps was obvious, e.g. comprising a series of routine tests and evaluations by the Skilled Team.
- In this case, there were accusations of hindsight from both sides. It is axiomatic that hindsight must be eliminated, but hindsight can infect both sides of the analysis. It is naturally critical that the obviousness attack must not be influenced or tainted by hindsight, but equally, hindsight must not infect the response to that attack. Hindsight can infect the response to an obviousness case if, for example, a witness (knowing of the ‘target’) appears to be looking for ways to avoid taking a particular step or making a particular choice towards the target, when the Court assesses that the Skilled Person would consider the step or choice differently and, having considered it dispassionately, would decide to take the step or choice.
- I mention this briefly because of certain cross-examination of Professor Buckton which was sought to be justified on the basis that the Professor had raised the competitive environment in his evidence. One of the pieces of prior art which was dropped was called Kumar which disclosed BAY-43-9006 and its chemical structure in a description of the competitive landscape relating to protein kinase inhibitors, which the Professor considered would be of interest to the wider skilled team. He pointed out that drugs targeted against protein kinases (such as sorafenib) were a new class of agents, the first of which, imatinib, had been launched in the US in May 2001 and other drugs which targeted protein kinases were undergoing clinical trials.
- Based on this mention of the competitive environment in Kumar, counsel suggested to Professor Buckton that if the Skilled Team were really thinking about the competitive environment they would observe that sorafenib was already in Phase 1 clinical trials, so there would be little technical or commercial driver to reformulate it. The purpose of the question was teased out by the Professor: ‘you were saying if Bayer already had this drug, you would not be looking to develop it?, to which Counsel answered ‘Correct’, the unspoken assumption being because Bayer would have a patent on it. Fortunately, this did not put the Professor off, since he stated his understanding that ‘the exercise for the court to consider [is] what you would do…. excluding that IP protection that would prevent you from doing it’, whereupon Counsel swiftly moved on.
- The Professor was right that the issue for consideration is what the notional skilled team would do without invention, having read the particular piece of prior art, without regard to the fact or possibility of patent protection which in the real world would or might prevent commercialisation of what the skilled team might develop.
- Lyons is an article about sorafenib and its utility in cancer therapy published in Endocrine-Related Cancer. The authors are identified as from Onyx, Bayer and Chiron. Its title is ‘Discovery of a novel Raf kinase inhibitor’. As reported in the Abstract, Raf kinase had been identified as a target for therapeutics with selective anti-tumour activity. It was not in dispute that Lyons was made available to the public on or before the priority date of the Patent.
- Lyons begins with a discussion of the relevant scientific background, namely, Ras epidemiology, Ras signal transduction, and validation of Raf kinase as a target in cancer before a section on the “Discovery of BAY 43-9006, a specific Raf kinase inhibitor”. The drug discovery program that led to the identification of BAY 43-9006 as a candidate for clinical development is described in this section together with its in vivo characterisation.
- The “Discovery of BAY 43-9006” section concludes by reporting that clinical testing of oral tablets of BAY 43-9006 in cancer patients commenced in July 2000. It reports that clinical testing of oral tablets of the drug in cancer patients was well tolerated; it has a relatively long terminal half-life of 35 hours in humans with preliminary clinical data being ‘encouraging’ because at least 37% of patients in this initial study had stable disease lasting longer than 12 weeks.
- The ‘Conclusion’ contains this passage:
‘BAY 43-9006 is an orally available potent inhibitor of Raf kinase with significant activity in four different human tumour types including colon, pancreatic, lung and ovarian cancers. Tumour growth was potently suppressed when BAY 43-9006 was dosed for 14 days, and this tumour suppression was maintained as long as dosing was continued. BAY 43-9006 also demonstrated significant anti-tumour activity against larger (400 mg - 1g) colon or ovarian tumours, with some regressions during the dosing period observed. These data suggest that BAY 43-9006 may have potential clinically as a cancer therapeutic with a novel mechanism of action.’
- As Professor Frijlink said, Lyons as a document would have been of more interest to a clinician than a formulator. However, the Lyons paper would have sparked the interest of medicinal chemists working in this area.
- Thus, as Professor Buckton explained, the Skilled Formulator would take comfort from the disclosure in Lyons that it was possible to formulate sorafenib as an oral tablet. Although the BAY code specifically refers to the free base, the Skilled Formulator would not have concluded that Lyons was disclosing that the free base had been used. S/he would have thought that sorafenib could have been formulated as the free base or a salt.
- Bayer pointed out that Lyons contains no direction to the skilled person to tosylate as the appropriate salt of sorafenib to use. As a result, Bayer submits that Teva’s case is essentially a CGK-alone case. They remind me of the warning by Birss J. in Accord v Medac, in this passage:
‘Obviousness over common general knowledge alone
119. I am not satisfied that the invention is obvious based on the common general knowledge alone attack relied on by Accord. That is because the relevant common general knowledge must be that in the UK (see Arnold J in Generics (UK) v Warner Lambert [2015] EWHC 2548 (Pat) at paragraphs 123-124) and in the UK the skilled clinician was unaware as a matter of common general knowledge what concentrations were being administered subcutaneously to patients and was unaware, without prompting, of any particular issue of pain arising from the subcutaneous administration. There is therefore nothing to provide an impetus to the skilled team to think about the issue at all.
120. Before leaving the argument based on common general knowledge alone, I will mention the words of Floyd J (as he then was) in ratiopharm v Napp [2008] EWHC 3070 (Pat) at paragraphs 155-159 and in particular the passage at paragraph 158 which warns that such attacks need to be scrutinised with care since they can be favoured by parties because the starting point is not obviously encumbered by inconvenient details of the kind found in documentary disclosures. I respectfully agree with Floyd J. Since it seems to me that this case provides a good example of the problems identified in ratiopharm I will add a few words of my own.
121. Normally the person attacking validity will rely on a particular concrete document or well defined prior use as a starting point. The fact that such a concrete item of prior art may be part of the common general knowledge is not the point. That is different from an attack based on common general knowledge alone.
122. Many inventions involve a combination of known features. However a combination of features, all of which individually were common general knowledge, can give rise to a valid patent claim if that combination is new and non-obvious. Patent trials are inevitably ex post facto and a key problem is to identify and avoid hindsight. Combinations of features can pose a particularly acute hindsight problem. The thing about concrete items of prior art, whether they are prior published documents or prior used products or processes, is that whatever combination of features that concrete prior art consists of, is not one which was created with hindsight knowledge of the invention.
123. The problem with arguments over common general knowledge alone is that the combination of features relied on is always and necessarily one created with hindsight knowledge of the invention, and worse, is one which the person attacking validity has not been able to find as a pre-existing combination in the concrete prior art. If they had they would have relied on that concrete prior art. Either the combination has not been made in the concrete prior art at all or it only appears with additional inconvenient details. If an invention is not obvious over the concrete prior art which is relied on, the court is entitled to be sceptical that an argument that it is nevertheless obvious over common general knowledge alone is correct.
124. The problem is illustrated in this case. Sometimes an invention belongs to a field which is not well documented but in this case Accord did not lack possible starting points. It has pleaded two documents and could easily have pleaded others, such as the existing SmPCs for subcutaneous methotrexate. However the documents contain what might have been thought of as "inconvenient" details. ……
… To invent as a starting point in the prior art an amalgam of the best bits of the two cited documents while leaving out the inconvenient aspects, which is in effect what the argument was, created a combination which did not hitherto exist.’
- Although I entirely agree with those observations, in my view they are not apposite in this case. Lyons does not contain any ‘inconvenient’ details. It discloses that a chemical entity identified by the BAY code showed promising therapeutic effect against four types of cancer. Then the question is: what would the Skilled Team do next?
- It is true that Lyons contains no direction to use tosylate. Equally it contains no direction to use either any salt of sorafenib or the free base - Lyons is deliberately uninformative as to the form of BAY 43-9006 actually administered.
- This is not a CGK-alone case. The starting point is Lyons. I find that Lyons provided the Skilled Team with a strong (but not irresistible) motivation to investigate this chemical entity with a view to identifying a formulated drug, preferably for oral administration to humans. The point is that from the starting point of Lyons, the Skilled Team (and the Skilled Formulator in particular) had their CGK routine tests, considerations and analysis to work with.
- The following points did not appear to me to be in dispute but in any event, I find the Skilled Team, having read Lyons, would take the following steps:
i) First, since Lyons reported initial results from Phase 1 clinical trials of BAY 43-9006, they would follow up relevant references including Riedl and Strumberg. The Riedl reference gave the Skilled Team the chemical formula for sorafenib. The Strumberg reference would have been followed up because it reported on pharmacokinetic aspects of the Phase 1 clinical trial. It revealed the dose range of between 50-400mg.
ii) Second, the medicinal chemists in the team would be tasked with finding a way to make the sorafenib free base. They would be able to make it (as Bayer admitted in correspondence), albeit initially in relatively small quantities.
iii) Third, I believe it was common ground that the Skilled Formulator would then seek to characterise the sorafenib free base.
iv) The Skilled Formulator would then find that the sorafenib free base was (a) weakly basic; (b) largely insoluble with a predicted and experimental solubility of 0.00171 mg/mL in water (the experts agreed this was a very low solubility); (c) with a calculated pKa in the range of 2.03-4.5 and 11.55-11.84. The two pKa ranges reflect the fact that sorafenib has both basic and acidic characteristics, but I am satisfied that the Skilled Formulator would decide to focus on its ability as a weak base to form salts, not least because (a) as Professor Frijlink pointed out, Aulton stated that drugs with a pKa of over 10 would not be suitable for salt selection and (b) the aim is to improve solubility at a pH found in the gastro-intestinal tract.
v) Professor Frijlink was keen to emphasise the very low solubility of the free base, in particular in relation to a contemplated high dose (he focussed on the highest reported dose of 400mg). He opined that the formulator would have considered this to be an extremely challenging project, especially taking into account a high contemplated dose. I am of the view that there was a degree of exaggeration here. Although Professor Buckton acknowledged that the free base had a very low solubility, his evidence (which I accept) was that by the Priority Date, working with NCEs with very low solubility was neither unusual nor unexpected for the Skilled Formulator, was something s/he would deal with day in/day out and one of the most common considerations faced.
vi) Bayer placed considerable reliance on a suggestion in Bastin that ‘aqueous solubilities in the range 0.1-1.0 mg/ml will normally be sufficient to satisfy the dissolution requirements for standard, solid oral dosage forms of drugs with good to moderate potency’. Whilst this range might be ideal, Professor Frijlink did not suggest that the low solubility of sorafenib would put off the Skilled Formulator from even attempting a salt screen.
vii) Based on these characteristics, the Skilled Formulator would decide to carry out a salt screen, in the expectation that they would find a sorafenib salt which would have improved characteristics and in particular improved solubility. Again, s/he would be encouraged by the knowledge from Lyons that the drug was administered orally in tablet form, and that form gave sufficient bioavailability (itself reflecting both solubility and permeability).
viii) Solubility would not have been the only concern. For the reason mentioned in paragraph 74 above, stability would also have been on the radar.
ix) The Skilled Formulator would know from the pKa values of sorafenib that, in order to form a suitable salt, s/he would need an acid with a low pKa. The Skilled Formulator would also know that pKa was not the only factor, but one which needed to be investigated (via different salts) to achieve a suitable balance of properties.
x) It is clear that different real-life teams would approach the salt screen in different ways. One group might just make and test the hydrochloride salt first, on the basis that the hydrochloride salt was the most common salt used. However, I find it is more likely that the Skilled Team would decide to make and test a number of salts simultaneously. Different teams might make and test salts in a number of tiers, with different numbers of salts in each tier. Professor Frijlink expressed the view that a standard salt screen at the priority date would contain around 4-5 salts, based on balancing a good probability of some salts forming successfully against conserving the limited amount of drug material available and keeping costs relatively low. In cross-examination, he accepted that if a first tier of 4-5 does not provide a candidate promising enough to take forward into the full formulation stage then the Skilled Formulator would try another 5 and possibly a further 5 beyond that. I think he was prepared to accept as many as 15 salts but only on the basis that the Skilled Formulator would only include the tosylate salt if s/he was ‘desperate’.
- As Teva submitted, the reason for undertaking a salt screen is because it is not possible to have an expectation that any given salt will have a particular set of characteristics. Some predictions or trends are mentioned in the textbooks, but, as I understand matters, it is precisely because the Skilled Formulator is dealing with a new chemical entity, that s/he has to select, make and test certain salts of that NCE in order to find which exhibits improved or acceptable characteristics e.g. of solubility and stability.
- It is at this stage that the critical dispute in this case arises. Although, as I have mentioned, much of Bayer’s closing submissions were aimed at the wrong target (i.e. claim 12 as proposed to be amended), the dispute really turns on whether it was obvious to include the tosylate salt in a salt screen. If it was, then the Skilled Formulator would ask the medicinal chemists to make the tosylate salt. They would be able to make tosylate sorafenib and that falls within claim 12.
- In their reports, both experts tackled the topic of salt selection in two stages. First, each set out some general considerations for low solubility drugs, albeit in differing degrees of detail. Although these included the points reflected in the Statement of Agreed CGK, each went further in various (and slightly different) respects. Second, each then considered what salts the Skilled Formulator would include in a salt screen for sorafenib. It would be fair to say that the general considerations each identified were, to varying degrees, influenced by the task in the second stage and it is not always possible to draw a clear line between the two stages.
- As I have indicated, the experts were agreed that a salt screen was a standard way to try to identify a salt of the NCE which had improved solubility. Professor Frijlink said a formulator would consider the properties of the drug compound under consideration and select salts they consider would be a good match. He emphasised that the formulator could not predict which, if any, salts might successfully be made nor what their properties might be but when selecting salts, he was of the view the formulator would consider a number of factors and he singled out the following five, to which I have added brief observations
i) First, the safety and usage of the counterion. He was of the view that the formulator would want to use counterions which had previously been used in approved clinical products, preferably in a number of major markets. As far as I could detect, this factor was solely about previous usage.
ii) Second, the pKa.
iii) Third, the anticipated indications of the candidate drug i.e. whether there were safety issues. Neither expert drew attention to any, although the Skilled Formulator would naturally be on the lookout for any toxicity issues which might arise.
iv) Fourth, the anticipated route of administration. It was common ground that the Skilled Formulator would aim for oral administration for its ease and convenience before considering other routes.
v) Fifth, the intended dosage form and dose. Again it was common ground that the Skilled Formulator would aim for a tablet form. S/he already had the anticipated dose of 50-400mg from the Strumberg paper, although I note that Professor Frijlink seemed to assume that the necessary dose was 400mg.
- He expected a standard salt screen to contain around 4-5 salts. Hydrochloride would be the first on the list, but he acknowledged in his written evidence that the formulator would have in mind the possible occurrence of a common ion effect, as well as the chance of a counterion exchange in the stomach even where an alternative salt was administered. In his oral evidence, although he acknowledged that the effect was widely discussed in all the textbooks, he suggested the phenomenon was ‘very rare’. This seemed to me to be a significant hardening of his view on the common ion effect. If it really was ‘very rare’ one would expect the textbooks to say so.
- Professor Buckton’s general considerations were focussed on dealing with an NCE with very low solubility. He was clear that the primary factor in deciding which counterions to include in a salt screen was the pKa of the ionisable group because this would largely govern the choice of salts available. After that, other considerations would depend on the properties of the underlying NCE and any issues the formulator was trying to address.
- In order to form a salt, Professor Buckton stated that the general rule was that the pKa of the acid and base should differ by at least two and preferably three pKa units. This was reflected in passages he cited from Bastin and Berge. Then, he said that the Skilled Formulator developing an oral dosage form would ordinarily want to use a counterion which has been previously approved by the regulatory authorities. He pointed out that these considerations mean that there was a limited list of counterions to select from, especially if the NCE has a low pKa. In addition, for weak bases with a pKa of less than about 3, he stated that the salts formed can have poor physical stability in the solid state and can easily disproportionate back to the free form with atmospheric moisture or when in contact with excipients in a drug product. Even then, salt formation was still the first choice for improving solubility, albeit that a greater focus is placed on the pKa difference in order to improve stability.
- So, Professor Buckton opined, in screening for an oral dosage form of an NCE with very low solubility, the Skilled Formulator would first identify acids with suitable pKa values which were already in use. Common acids were identified in books such as Aulton and Bighley and in journal articles such as Berge, Gould and Bastin and he said that the acids mentioned in those texts were indicative of the counterions commonly used by the Skilled Formulator in a salt screen.
- In cross-examination, he was challenged with extracts from his reports in other pieces of litigation where he was content with a difference of 2 pKa units between acid and base. These challenges seemed to me to go nowhere for three reasons: first, because, as Professor Buckton explained, those reports were considering different circumstances; second, because Professor Frijlink agreed that the skilled formulator would want a difference of 2 units, although on occasion he suggested a difference of 1 would do; third, because of Professor Buckton’s point that stability was likely to be improved by a greater difference in pKa units.
- On that third point, in cross-examination Professor Buckton gave a clear explanation of the technical reasons behind his ‘at least 2 and preferably 3’ range, by reference to the pKa range for sorafenib (2.03-4.5). In essence, he explained that if the pKa was at the lower end of the range, the acid has to have a pKa which is much lower because ‘we are in the region where it is going to be very difficult to get a salt to hold together. If we are at the 4.5 pKa… that becomes a little less critical.’ Hence he explained that if the drug substance has a pKa of around 6 or 7, then he would use his general rule of 2 as a difference; if around 4.5, he would be looking for an acid with a pKa difference of 2.5; if around 2, then he would be looking for a pKa difference of 3; and the purpose of the greater difference at the lower pKa values was to achieve a stable salt.
- Furthermore, as I indicated, Professor Buckton’s range was supported by passages in both Bastin: ‘for the formation of a stable salt, it is widely accepted that there should be a minimum difference of about 3 units between the pKa value of the group and that of its counterion, especially where the drug substance is a particularly weak acid or base’ and Berge: ‘particularly important is the relative strength of the acid or base…these factors determine whether or not formation occurs and are a measure of the stability of the resulting salt’.
- Before he came to consider sorafenib in particular, Professor Buckton presented a list of acids which lower melting point or have strong interactions with water, which the Skilled Formulator would have in mind when seeking to improve solubility. He based this list on Gould. He noted that Gould and Bighley present a stepwise approach to salt selection, but he considered that theoretical, and in the real world different counterions would be screened for salt selection simultaneously in order to save time overall. Although he was criticised for his ‘real world’ approach, I do not consider the criticism was justified. The ‘stepwise’ or ‘decision tree’ approaches were being advocated, in my view, to increase efficiency. So, instead of a formulator drawing up a long ‘laundry list’ of possible salts, these texts suggested that salts should be selected based on some assessment of the available guidance. This was what both experts suggested and did in their evidence, albeit with differing degrees of emphasis. So, Professor Buckton’s introduction to and his general list read as follows:
These papers therefore give overall guidance for acids and bases that would be considered as possible counterions as well as trends in the expected properties of salts when they are made. The guidance shows that some acids will make a salt less soluble, and others will be expected to enhance solubility of a poorly soluble basic compound. For example, solubility can be enhanced by using acids which lower melting point or have strong interactions with water41. Examples of acids which can improve solubility are the mineral acids (e.g. hydrochloride, hydrobromide, sulfate, phosphate), methane sulfonic acid (mesylate), maleic acid (maleate), p-toluene sulfonic acid (tosylate), 2-ethane sulfonic acid (edisylate), acetic acid (acetate), succinic acid (succinate), lactic acid (lactate) and other hydroxy carboxylic acids42. In contrast, hydrophobic counter ions e.g. pamoate would be expected to reduce solubility43. Whilst the expectations and general trends exist, numerical values cannot be obtained prior to the salt being formed44 and, therefore, salt selection is a matter of routine testing of multiple salts in order to obtain salts with the optimal balance of properties. The selection of acids with which to make salts of an API will be guided by the concepts in these references, but the salts will need to be made and tested to ascertain which one(s) will have the best overall properties.
- Two important footnotes in that passage are:
i) fn42: ‘See Gould (GB-07) teaching generally that mineral acids can improve solubility, as do those acids listed in the first half of series 1 in his Table 3, and progressing into series 2 in his Table 3.’ As Professor Frijlink correctly discerned, this footnote was based on this passage in Gould on p213: ‘To increase aqueous solubility for basic drugs it would seem appropriate to proceed half way up series (1) (i.e. before crystal forces dominate the solubility) or to extend into the hydroxy acids series (2).’
ii) fn43: ‘See Gould (GB-07) "the latter portion of series (1) would also serve for movement to potentially more insoluble salt forms". Pamoate is the last member of "series 1" of Table 3 on p212.’
- Gould’s Table 3 (as helpfully annotated by Professor Frijlink) looks like this:
- Professor Buckton said the hydrochloride salt was the most frequently used acid addition salt, but after that all other acid addition salt formers have a much lower percentage usage and there was generally no clear order of priority after hydrochloride. He, too, drew attention to the possibility of a common ion effect with hydrochloride. He said, relying on Aulton, that if a common ion effect is observed, then other salt forms are indicated e.g. tosylate, mesylate etc.
- In his reply report, Professor Frijlink divided Professor Buckton’s general list (contained in the quote in paragraph 139 above) into three parts because he wanted to explain the extent of his disagreement. Professor Frijlink did not agree that all of the counterions in the list would be understood by the formulator as improving solubility, either as part of their CGK or based on Gould. Professor Frijlink agreed regarding the mineral acids (Part A), and with the series 2 counterions (his part C) with the qualification that the skilled formulator would be more familiar with some than others: ‘tartrate, acetate, citrate and succinate were more often used in a pharmaceutical context compared, for example, to edisylate or lactate.’
- Professor Frijlink’s Part B concerned ‘methane sulfonic acid (mesylate), maleic acid (maleate), p-toluene sulfonic acid (tosylate)’. He agreed with mesylate and with maleate (on the basis it would be ‘less obvious’ to choose than mesylate) but he disagreed as regards tosylate, giving two reasons:
i) First, he regarded Gould’s reference to ‘half way up’ series 1 as being ‘not very clear’;
ii) Second, he took the view that the formulator would consider that tosylate could be a counterion where ‘crystal forces dominate the solubility’ and also increase hydrophobicity and stability. In support of this point, Professor Frijlink relied on this passage from p 485 of Bighley:
"Sulfonic acids are being used more and more for various drug candidates because they can be manipulated to influence dissolution rate and reactivity. For instance, the mesylate salt is frequently highly soluble and therefore dissolves quickly. On the other hand, the hydrophobicity of the sulfonate can be increased by the incorporation of longer carbon chains (esylate, edisylate, isethionate, besylate, tosylate, pamoate (embonate), napsylate, xinafoate, and estolate)."
- Professor Frijlink pointed out that whilst the mesylate is described as ‘frequently highly soluble’, tosylate is presented further down the list of increasing hydrophobicity, which the formulator would associate with reducing solubility.
- In his first report, Professor Frijlink considered that the tosylate salt would not be included in an initial salt screen for a counterion for sorafenib. By ‘initial salt screen’ it is evident he meant a screen of 4-5 salts. He said he did not personally recall ever coming across it and he never used it in a salt screen at Solvay. He said it was a strong acid but hardly used at all. He acknowledged it was present in some counterion tables (e.g. in Bastin, the 1st edition of Aulton and in Bighley). He considered that the formulator would not have been familiar with this counterion in a pharmaceutical context. For all those reasons he did not consider it was an obvious choice to consider.
- Instead, Professor Frijlink considered the formulator would select the following counterions to screen for sorafenib, for the reasons he stated:
i) Hydrochloride –it is a strong inorganic acid, by far the most common counterion for basic drugs and included in almost all salt screens;
ii) Sulphate - another strong inorganic acid, well known and the second most common counterion;
iii) Phosphate - a well-known and established salt, a medium strength inorganic acid and with reasonable usage;
iv) Mesylate - a strong sulfonic acid with reasonable usage.
- It is apparent from his reasons that he was relying on the strength of the acid (i.e. its pKa) but primarily on the degree to which the counterion had featured in marketed drugs (consistently with this being the first factor he identified in the first stage). In his second report, contemplating a slightly wider screen perhaps, but mostly in response to the range of counterions mentioned in Professor Buckton’s first report, he considered that the formulator would select, in addition, hydrobromide, maleate, tartrate and citrate, on the basis that they have pKas in the range of 2-3, even though the pKa of hydrobromide is much lower (<-6) and, as Teva submitted, hydrobromide does not feature in Table 13.4 of Aulton. I note from Table 13.4 in Aulton that the lowest pKas given for these counterions are maleate: 1.92; tartrate: 3.00; citrate: 3.13. As Teva pointed out, these pKas are all considerably higher than tosylate: -1.34 and mesylate: -1.20, but it is true that Table 13.4 gives the lowest % usage of 0.1 to tosylate.
- For his part, Professor Buckton focussed initially on acids with a low pKa. He pointed out that the Skilled Formulator would be aware that, when working with APIs with a pKa of less than about 3, sometimes the resulting salts can be less than optimally stable. However, when the pKa value was only a small amount less than 3, there would be an expectation that stable salts suitable for oral administration could be formed if matched with a strong enough acid counterion. For these reasons, he stated that the Skilled Formulator would want to use an acid with a pKa value which is at least 2 and preferably 3 pKa units lower than that of sorafenib.
- With that approach in mind, Professor Buckton’s Skilled Formulator would select acids with a suitable pKa value from the standard lists contained in the textbooks, with a view to improving solubility. His reasoning was simple: try the mineral acids (hydrochloride: pKa -6.10, sulphate: -3) but familiarity with possible common ion effects associated with hydrochloride salts would motivate the inclusion of alternative acids with low pKa including sulfonic acids (tosylate: -1.34, and mesylate -1.2, as suggested by Aulton) and carboxylic acids (e.g. tartrate: 3, citrate: 3.13). Professor Buckton was of the view that it would not be inventive to include any of these salts in a salt screen, and that the primary factor in deciding which counterions to include would be the pKa of the ionisable group for the reasons he had already explained. In this regard, I note that Professor Frijlink agreed that the pKa of the tosylate counterion made it an attractive pKa from the perspective of making a salt with a weak base.
- In view of his reliance on Aulton, counsel for Bayer suggested to Professor Buckton that he only pointed to the passage in Aulton on the common ion effect with the benefit of hindsight. The Professor disagreed, and so do I. Counsel further suggested that the entirety of Aulton was not CGK. Professor Buckton responded by saying he thought the chapter in Aulton was CGK, on the basis that Aulton was ‘a basic undergraduate textbook’ and he would expect his undergraduate students to know what it says in there, the same for people working in the pharmaceutical industry. I found his explanation convincing.
- Counsel then sought to divert attention away from Aulton by pointing out that Bighley discussed the common ion effect but suggested a different solution - the use of a hydroxy counterion, not a sulfonate. In response, Professor Buckton pointed out that the use of a hydroxy acid might well not be suitable in this instance because they have (relatively) high pKas, although I note that he was of the view that the Skilled Formulator would be likely to include 2 hydroxy acids in his salt screen.
- Although the salt screen would include the hydrochloride salt (probably as the first on the list), it is a fact that the hydrochloride salt of sorafenib did not emerge as the administered drug and this is likely to have been the result of the common ion effect. Hence it is likely that the Skilled Formulator would have encountered the common ion effect or some other reason to exclude the hydrochloride salt.
- Bayer made a series of submissions on the common ion effect:
i) That it was in fact a very rare phenomenon (based on some of Professor Frijlink’s answers in cross-examination), a point reinforced by the fact that such a high proportion of drugs are formulated as hydrochloride salts. However, as Bayer acknowledged, all the major texts drew attention to the common ion effect, and I find it would be on the Skilled Team’s radar because of the inclusion of the hydrochloride salt. Furthermore, since an estimated 45% of drugs in the 1974 survey used the hydrochloride counterion, that means that the majority of marketed drugs did not, perhaps an indication that the common ion effect was not as rare as Bayer contended.
ii) That Professor Buckton ‘simply junked Teva’s case on the common ion effect’. This submission was founded on this exchange in the cross-examination of Professor Buckton:
5 Q. I suggest to you that the skilled formulator would not
6 automatically think of mesylate or tosylate, even if the
7 common ion effect arose?
8 A. I would agree.
However, in my judgment, neither that answer nor any other ‘junked’ the relevance of the common ion effect. Unpacking that exchange, Professor Buckton was agreeing that if the common ion effect arose, the hydrochloride salt would not be suitable, so the Skilled Team would have to look at other salts. That leaves the question of ‘which other salts’. Professor Buckton was realistically accepting that the Skilled Team would not ‘automatically’ think of mesylate or tosylate. But that does not rule out that they would, after further thought, select mesylate or tosylate for testing in their salt screen. This is a point he explained in his answer, the full text of which was as follows:
8 A. I would agree. As I said yesterday, I do not think you would
9 do these things sequentially anyway. I think you would make
10 the hydrochloride as part of a larger number of salts and you
11 would test the properties of those salts, and on the back of
12 that, if the hydrochloride in your in vitro testing, or if you
13 ended up doing a dog study, had a common ion effect problem,
14 you would select from one of the other salts that you have
15 made. So I would not suggest that you do the hydrochloride
16 and on the back of a common ion effect go and make the
17 tosylate, but rather I would suggest that tosylate is a
18 sensible alternative, particularly for low pKa drugs.
Reverting to the relevance of the common ion effect, I note that Professor Frijlink in his first report acknowledged that the Skilled Formulator would have had in mind the possible occurrence of the common ion effect (and it was part of the Agreed CGK that the Skilled Team would know of the common ion effect), as well as the chance of a counterion exchange in the stomach even if an alternative salt (i.e. not Cl- ) was administered. I have already referred to the hardening of Professor Frijlink’s view of the common ion effect in his oral evidence.
iii) That, if the common ion effect arose, tosylate was not put forward as the universal prescription. Bayer pointed out, correctly, that tosylate was mentioned in Aulton, and submitted that Bighley, for example, suggested using hydroxy acids, if the common ion effect arose. Bighley considers the common ion effect a number of times in relation to different properties under consideration. Under the heading of Solubility and the sub-heading of Common Ion effect, I was unable to find any recommendation to use hydroxy acids, but I accept that if the common ion effect arose, the Skilled Formulator would consider other acids, based on his or her pKa considerations.
iv) That Professor Frijlink gave reasons why, having read the relevant passage in Aulton, the Skilled Team would still be put off selecting tosylate for their salt screen. I deal with these reasons below.
- I will not set out the entire passage in Aulton under the sub-heading of ‘Common ion effect’ but Aulton points out that ‘Hydrochloride salts often exhibit suboptimal solubility in gastric juice due to the abundance of Cl- ions. …. Other counterions, other than Cl-, such as nitrate, sulphate and phosphate, have also been implicated.’ After a short passage detailing how to identify a common ion interaction, the text continues as follows:
A common ion effect with Cl- will result in significantly reduced IDR [intrinsic dissolution rate] in the presence of sodium chloride. Other salt forms are then indicated, e.g. sulphate, tosylate, mesylate etc., but the parent molecule may well still remain sensitive to Cl- and solubilities will be suppressed in the presence of saline although not to the same extent since Cl- Is not involved in the dissolving microenvironment. ….
Where a hydrochloride salt exhibits suboptimal solubility then the next logical choice is probably a salt of toluene sulphonic acid (tosylate: pKa - 1.34). Mesylate, napsylate, besylate and maleate salts offer progressively more weaker acid alternatives (Table 13.4). ….
- In relation to these passages from Aulton (which appear in both editions), Bayer relied on these answers from Professor Frijlink (T2/p291):
14 Q. That is reasonable guidance for a skilled person to follow;
15 correct?
16 A. Well, not completely. I think that the toluene should not
17 have been that high up the list because you know you have
18 these planar things, and the skilled formulator would know
19 that, and this is something where I feel that it would be
20 better to start with mesylate. The (unclear) sulfonic acid
21 dose molecules are much more appropriate there than the ones
22 having the benzene or the naphthalene rings in them. So, it
23 is written there, but the skilled formulator would know that
24 there is a certain risk in going that way.
25 Q. What I want to suggest to you is that you are rather
2 over-thinking this issue, from your professorial standpoint,
3 and it is not a criticism of you. A skilled formulator is
4 notionally uninventive. He would not be thinking, with the
5 level of sophistication that you are bringing to this, to say,
6 "There is a benzene ring here. It might cause problems in
7 relation to solubility". This [I interpolate that in context ‘This’ was a reference to the passage in Aulton] reflects the thinking of a
8 notional skilled person; correct?
9 A. No. I think when I was working at Solvay and we did these
10 salt selection screens we were aware of the fact that these
11 benzene rings in the counterions had their problems because of
12 the crystal formation. We were really, really aware of that.
13 Q. That was your ----
14 A. No, that was not something special. I think most industries
15 had that.
- Bayer also submitted that once the skilled formulator had taken into account solubility as well as pKa, if they encountered the common ion effect then the non-aromatic sulphonic acids would be much more sensible to pursue, relying on this answer from Professor Frijlink (T2/p292):
24 Q. Okay, but even suppose that the skilled formulator thought
25 that, there would be a rationale for including the tosylate on
2 the list, precisely because it would not be possible to tell,
3 in advance, whether or not this would actually be an obstacle
4 to salt formulation?
5 A. You could add it, but with a very low expectation of success.
6 If your primary objective is to increase solubility, which it
7 is in many cases of salt formation, then toluene is a poor
8 choice. Of course, in certain states of something you may add
9 it because there is a little chance, but that is all you can
10 do, and the expectation of success is really minimal. You
11 would understand, and you would also understand why. It is
12 because these benzene rings are in there. You would better do
13 edisylate or esylate. They have a much larger chance to stand
14 success. They are also sulfonic acids.
- Although there was scientific validity in Professor Frijlink’s point in the sense that if one digs around in Gould, for example, one can find some support for his point on the influence of the benzene ring in the tosylate counterion (and related points made in his second report regarding aryl groups), in my judgment, Professor Frijlink’s views on the suitability of the tosylate counterion for inclusion in the salt screen for sorafenib departed from those of the Skilled Formulator for a number of reasons, as follows.
- First, he was prepared to disagree with the suggestion made in Aulton (both editions) that if the common ion effect arose the formulator should try certain other counterions, of which tosylate was either first or second (after sulphate). His disagreement was limited to the tosylate counterion. Furthermore, he said the Skilled Formulator would disagree with that suggestion and I find that would not have been the case. I found his view surprising and at the conclusion of his evidence, I asked Professor Frijlink whether a formulator who followed such suggestions would get ‘fired or criticised’ for doing so. Perhaps not surprisingly, he answered in the negative. I find the Skilled Formulator would have noted and taken up Aulton’s suggestion.
- Second, I found it somewhat illogical that the Professor’s Skilled Formulator would have the frequency of use of the counterion in question as his primary factor in his selection criteria. Once the hydrochloride and sulphate counterions are selected, all the others have low percentage usage figures. Furthermore, frequency of use in other drugs (e.g. in Aulton 1st, Table 13.4), when the data was out of date and did not include the more recent drug candidates with very low solubility, tells the Skilled Formulator very little, if anything, about the suitability of those counterions for selection to attempt to deal with whatever problem(s) are discovered with a particular drug candidate. For the avoidance of doubt, I find that the Skilled Formulator would not have gone searching to see if anyone had published more up to date data. I note that Professor Frijlink identified a paper published in 2007 (Paulekuhn) and he discussed the paper in some detail but obviously the post-priority data contained in Paulekuhn would not have been available to the Skilled Formulator. In any event, Gould lists 4 uses for the tosylate counterion, albeit these were probably not FDA approved drugs.
- It is true that the hydrochloride counterion was well out in front in the usage stakes and that is why it would always be tried, but I was struck by the fact that of the counterions which Professor Frijlink would include in a salt screen (a total of 8), with one exception (hydrobromide) they are a list of the 7 most frequently used counterions from Aulton’s Table 13.4: hydrochloride (43%), sulphate (7.5%); phosphate (3.2%), mesylate (2.0%), maleate (3.0%), tartrate (3.5%), citrate (3.0%), and this selection appears to have been made without regard to the pKa values.
- I find much more force in Professor Buckton’s primary factor: consideration of the pKa and the likely ability of the counterion to form a stable salt of sorafenib. With the range of pKa of sorafenib on which both experts were working (2.03-4.5) and the need for a suitable pKa difference (2-3), I found Professor Frijlink’s inclusion of maleate: 1.92; tartrate: 3.00; citrate: 3.13, inclusion of mesylate (-1.20) but exclusion of tosylate (-1.34) to be illogical. No reasons were put forward to favour one end of the pKa range for sorafenib or the other. Even if a pKa difference of one might have been considered to be suitable, it still does not make sense to exclude tosylate (which would appear to stand a good chance of forming a stable salt) but include tartrate and citrate (where the pKa numbers indicated a serious risk of not forming a stable salt at all).
- These points (the primary factor point, and consideration of the pKas) reflected, in my view, a more general tendency on Professor Frijlink’s part to look for reasons to exclude tosylate from the salt screen candidates (and with the benefit of hindsight) when the Skilled Formulator would be dispassionate and have no preconceptions. Other indicators of this tendency were:
i) He argued that the suggestion in Gould to go ‘halfway up series 1’ to improve solubility was ‘not very clear’. However, because the Skilled Formulator would consider this recommendation without any preconceptions, s/he would read ‘halfway up series 1’ as including tosylate. Although series 1 has 5 entries, with tosylate in the middle, the size of the counterions in entries 4 and 5 place tosylate in the first half (reading from the left). I find that the dispassionate Skilled Formulator would not have any problem or issue over Gould’s suggestion and would take it to include the tosylate counterion.
ii) He placed particular emphasis on the presence of the benzene ring (a point which manifested itself in his references to the aryl group and to toluene) and its effect in increasing the hydrophobicity of the salt and therefore being less suitable for increasing solubility compared with other counterions. When discussing Gould in his second report, he was at pains to stress ‘the formulator’s view of counterions with larger hydrophobic or aryl groups, which might contribute to a more stable crystal, potentially with a higher melting point and a lower aqueous solubility’ (my emphasis). Whilst I agree that the Skilled Formulator might know of the use of the pamoate counterion to make a salt more insoluble (to control absorption of a drug - see the end of series 1 in Gould and the specific discussion in Gould on p208), this effect is discussed in Gould and is apparent from the fact that the size of the counterion increases across series 1. There is reference in Gould to a study of aryl sulphonic acids being tested as salts to protect an easily hydrolysed base (xilobam) and mention of the aryl groups presenting a hydrophobic barrier to minimize hygroscopicity and dissolution in surface moisture, but the discussion is in the context of stability being a particular problem due to surface moisture. However, the detailed discussion in Gould (which I doubt was CGK) amongst the various pivots is then simplified into his recommendations for addressing specific issues (which I consider was CGK). In that context, under the heading of solubility, the Skilled Formulator finds the sentence quoted in 140.i) above. In my judgment, the Skilled Formulator who consulted Gould would apply his recommendations.
iii) In any event, as I indicated earlier, although solubility was the principal issue sought to be ameliorated by assessment of various salts in the salt screen, it was not the only issue and the Skilled Formulator would have also had at least a concern about stability. In this regard, I found a certain inconsistency between some of Professor Frijlink’s answers. At T2/p271 he was keen to stress that making the salt depended on two things: the pKa and the stability of the crystal - reflecting the common ground that the Skilled Formulator had to consider a balance of factors and properties. However, when it came to his view of the tosylate counterion as a potential candidate, he seemed almost to have dismissed stability as a consideration, focussing only on solubility (see e.g. the answer relied upon by Bayer, quoted in paragraph 157 above). The Skilled Formulator would have been seeking to strike some sort of balance between solubility and stability, albeit s/he would know they had to make and test salts to find that balance. If or to the extent that the Skilled Formulator did share some of Professor Frijlink’s concerns about the influence of the benzene ring, they would not, in my view, have put him or her off to the same extent as the Professor.
iv) At this point I mention that Counsel for Teva cross-examined Professor Frijlink by reference to a table (prepared for the purposes of his cross-examination) of properties of various counterions, the properties including pKa, molecular weight, solubility and melting point. The point of this part of the cross-examination was to establish that (a) the solubility of tosylate alone was favourable (not as soluble as mesylate, but more soluble than a number of Professor Frijlink’s other suggestions including maleate, tartrate and citrate, and, as the Professor accepted, the solubility of ‘pure tosylate’ was not much affected by the presence of the toluene group (which contains the benzene ring)); (b) the molecular weight of tosylate was less than that of citrate and would not have been ruled out on that basis, another point which the Professor accepted. I have not placed any weight on this evidence because it was not established whether the entries in the table which were the subject of these questions were CGK or would have been available at the Priority Date. In any event, this questioning was another level of detail beyond that which the Skilled Formulator would engage in.
v) On the benzene ring point, the Professor spoke (in his answer quoted in paragraph 156 above) of particular problems they had experienced at Solvay with ‘these benzene rings’. I do not consider this experience reflects that of the Skilled Formulator and it does not accurately reflect what the Skilled Formulator would glean from the standard textbooks and articles. If one has the issue of benzene rings already in mind, it is possible to find material in the CGK sources which reflect it, but that is approaching matters the wrong way round. In any event, none of the CGK sources present a red flag regarding the potential hydrophobicity of tosylate. The Skilled Formulator would understand it is all a question of degree.
- To be clear, the two particular passages relied upon by Bayer (and quoted above) are, I find, both examples from Professor Frijlink’s oral evidence where he was focussed on finding reasons not to include the tosylate salt in the salt screen, and was doing so with the benefit of hindsight.
- I emphasise these are all subtle points. As I have mentioned, there was scientific basis for the points made by the Professor and with his points in mind, as I said, it is certainly possible to identify passages in Gould (and Bighley) which support them. The issue, however, is whether the dispassionate Skilled Formulator would have focussed on or placed the same emphasis on these points as Professor Frijlink. I am satisfied that s/he would not. Overall, I do not consider that real-life teams would have agonised over or debated these points to the extent they were in this case. It is far more likely that they would have said to themselves ‘we cannot make any firm theoretical predictions, so let’s get on and make a selection of salts and test them.’
- In Bayer’s Closing Submissions, my attention was drawn to a number of Professor Buckton’s answers which support this point. For example:
i) ‘there are generalisations and trends. You cannot put numbers to it or solutions to it'.
ii) And:
8 Q. At best it is all about general trends and pivot points, but
9 in the end it is impossible to predict which salt would have
10 the optimum properties for use?
11 A. That is fair. That is fair.
12 Q. You cannot even predict if crystals will form for any
13 particular salt, can you, Professor?
14 A. No.
15 Q. Let alone whether a particular salt might improve solubility,
16 dissolution rate or bioavailability?
17 A. No. You can take the trends that we have just talked about,
18 but I cannot tell you the numbers I am going to get.
iii) Bayer also pointed out that Professor Buckton accepted that tosylate might be a counterion in which the crystal forces dominate the solubility considerations - he simply did not know, given the complexity of the chemicophysical picture.
- Such subtlety as there was in the evidence was lost by the time it came to Bayer’s closing submissions. To give two examples, Bayer submitted that:
i) ‘Professor Frijlink explained that the clearly hydrophobic, electron-rich, planar aromatic group on the tosylate moiety is a clear reason not to screen it for sorafenib’ and
ii) ‘….. tosylate just wouldn't get on the skilled formulator's radar: it is an obscure choice, advanced only by one book as a possible solution to a problem that does not appear to apply; and there are very good reasons to suppose it would do more harm than good, given general considerations about its aromatic structure.’
iii) Even if I had not rejected the evidential basis for these submissions, they still involved significant exaggeration. For example, Bayer were keen to stress the solubility range in Bastin, whilst conveniently forgetting that Bastin listed the tosylate counterion in his classification of ‘common pharmaceutical salts’. Bastin’s Table 1 classifies ‘common pharmaceutical salts’ by type. The inorganic acids (5 are listed) come first, followed by the sulfonic acids (6 are listed, of which tosylate is the fourth). It is simply incorrect to suggest that tosylate is advanced by only one book. In the main, all the CGK sources speak of sulfonic acids as potential counterion candidates. Whilst the large pamoate counterion is singled out as significantly reducing solubility where this is required, none of the CGK sources gave, in my view, anything like a ‘clear reason’ not to try it for sorafenib.
- Reverting to my findings on Professor Frijlink’s evidence, my conclusions above are reinforced somewhat by a passage at the conclusion of Professor Frijlink’s first report where he said he did not recall ever coming across tosylate and had never used it in a salt screen whilst at Solvay. He said he did not believe the formulator would have been familiar with the counterion in a pharmaceutical context. His later expressed views about the benzene/aryl/toluene group are not necessarily inconsistent with that, but if the formulator was not familiar with the tosylate counterion, it seems to me to be far less likely that s/he would have been focussing on the impact of the benzene ring to the extent that the Professor did.
- My conclusion is also reinforced by what I consider to be at least a contributory reason for the Professor’s ‘very low expectation of success’ in the answer quoted in paragraph 157 above. This emerged later in the Professor’s cross-examination and it concerns the solubility of sorafenib tosylate. This came into the case in the following way.
- Professor Buckton referred, in his first report, to a predicted figure for the solubility of sorafenib from a particular database of 0.00171mg/ml. Having read Professor Buckton’s first report, Professor Frijlink searched for properties of sorafenib on the internet and found a predicted figure for the solubility of sorafenib with the same value on DrugBank. He noted there was also a section on sorafenib tosylate, with a separate solubility figure, predicted using the same software of 0.00152 mg/ml. This is lower than the predicted solubility for the free base, but only slightly so.
- He noted there was no information on DrugBank as to what conditions were taken into account when making this prediction, in particular with respect to pH. The experts were agreed that any significant improvement in solubility would be expected to be achieved below the pKa of sorafenib. The Professor then went on in his reply report to say he was directed by Bayer’s solicitors to publicly available data on sorafenib tosylate in regulatory documents available the European Medicines Agency (EMA) and he quoted as follows:
‘The solubility of sorafenib tosylate ranges from 0.034mg/100ml at pH 1.0 to 0.013 mg/100ml at pH 4.5.’
- He noted this is a solubility of 0.00034mg/ml at pH 1, a figure which he said was ‘incredibly low, even lower than the modelled values on DrugBank for either sorafenib or sorafenib tosylate.’ He concluded: ‘The formulator’s expectation of success for developing a solid oral dosage form with any salt with aqueous solubility this low would be extremely low.’
- This solubility figure for sorafenib tosylate obviously struck the Professor as highly significant since, later in his cross-examination, he made the point numerous times that the tosylate made the solubility ‘ten times worse’ than that reported for the free base (in fact, based on those figures, it is almost exactly five times worse). On that basis he said it was completely counterintuitive to take the sorafenib tosylate salt through into the formulation stage to produce tablets for oral administration.
- A number of points arise:
i) First, in addition to some exaggeration, the Professor was not comparing like with like. The calculated figures suggested that the solubility of the tosylate was comparable with the free base. The figure derived from the EMA document was presumably a measured figure, but there was no measured figure for the free base.
ii) Second, but more importantly, in those answers, the Professor evidently had in mind the proposed amended claim. However, as regards Claim 12 in its unamended form, the Skilled Formulator would not know or be able to measure the solubility of sorafenib tosylate until after the team had made that substance and subjected it to the normal pre-formulation tests. So the quote from the EMA document was a piece of hindsight knowledge as regards claim 12 in its original form.
iii) If it had been clear from the Professor’s answers that he had striven to keep that piece of hindsight knowledge out of his mind when considering the prior stage of which salts to include in the salt screen, the answers relied upon by Bayer (as quoted above) might have had greater force. However, as I have already indicated, I formed the clear impression that Professor Frijlink was looking for reasons not to include the tosylate salt in the salt screen, and this was a further manifestation.
iv) The final point to note from the EMA document exhibited by Professor Frijlink is this sentence: ‘Due to the very low solubility of sorafenib in aqueous media, the tosylate salt was used in the drug product.’ This sentence implies (strongly, in my view) that, as a matter of scientific fact, the solubility of sorafenib tosylate was actually better than that of the free base or, at the very least, the combination of properties of sorafenib tosylate (e.g. solubility, stability, permeability etc) were an improvement on the free base. This sentence was neither commented on by Professor Frijlink in his second report, nor drawn to his attention in cross-examination.
- Overall, I conclude there was considerable force in Mr Alexander QC’s suggestion in his question, quoted above, that Professor Frijlink was ‘over-thinking’ the process which would have occurred in the notional Skilled Formulator’s mind. Furthermore, the Professor was bringing to bear more specialised knowledge which he had (e.g. regarding the impact of a benzene ring or his knowledge of the solubility figure for the tosylate salt) which was not CGK.
- Hindsight also featured heavily in a number of propositions which Counsel put to Professor Buckton in his cross-examination. A number of propositions were put with a view to ensuring that the tosylate salt was excluded from consideration in the salt screen. But, as Professor Buckton indicated in a number of his answers, you cannot predict accurately what is going to work (and not work) which is precisely why, in this semi-empirical field, one has to carry out a salt screen to find out.
- Bayer drew attention to the number of cases in which Professor Buckton had appeared for parties attacking a patent (some 25 which had reached court or deposition), but I note that Professor Frijlink had also appeared in a large number of cases of this type. The implication in Bayer’s point was that Professor Buckton (almost) always found a way to invalidate formulation patents in cases in which he appeared but he responded by indicating that was a generalisation. He also pointed out that he had held views that a patent was not obvious at all, but suspected that those ones did not reach trial because of his view. It was also suggested to him that, in some cases, he came to his opinion based on hindsight, but he responded that he hoped not, because he made every effort to exclude hindsight. These (and other) suggestions were made with a view to persuading me that Professor Buckton’s opinions in this case were driven by hindsight and/or some sort of animus against formulation patents.
- I reject all such suggestions. Particularly in his oral evidence I found Professor Buckton to be measured and dispassionate. He did not exaggerate and was prepared to agree with points which might have appeared to be contrary to his overall opinion. I am entirely satisfied he was doing his level best to assist the Court.
- In its closing submissions, Bayer invited consideration of a number of other questions, such as ‘why is tosylate so rare?’ but this is just the usage point in a different guise. As I indicated above, although Bayer made trenchant submissions on which salts to include in a salt screen, it also made some submissions which were aimed at the wrong target i.e. claim 12 as proposed to be amended. I have already mentioned how my raising of this point in closing submissions gave rise to the issue of construction of claim 12 which I have addressed above. Here I mention the final response from Mr Mitcheson QC on that point. Ultimately, Mr Mitcheson QC reserved his client’s right to bring back the amendment to the patent post-judgment. If such an application is made, it will be considered on its merits, but it does not deter me from the approach I have taken in this judgment nor my conclusions.
- With the above analysis in mind, I revert to the issue I identified in paragraph 129 above. I can state my conclusion succinctly. I find it was obvious for the Skilled Formulator to include the tosylate salt of sorafenib in his or her salt screen. S/he would have directed their medicinal chemists to make sorafenib tosylate and the other selected salts. Hence claim 12 was obvious.
- My reasons are set out at length in the analysis above, but in summary:
i) When deciding which salts to include in his or her salt screen, the Skilled Formulator would have consulted at least Aulton and one or more of the other publications I listed in paragraph 26 above, if not already familiar with the considerations I have identified as CGK above. I find that the primary factor in the salt selection process was the Skilled Formulator’s consideration of the pKa range of sorafenib and the pKas of the various counterions. Those considerations would have identified tosylate as an attractive candidate, as Professor Frijlink accepted.
ii) Professor Buckton accepted that this field was conservative and regard would be had to counterions which had featured in marketed drugs, but I do not believe this factor would dominate the selection process in the manner implied in Professor Frijlink’s evidence. Although Bayer sought to establish the 0.1% usage in Aulton (1st Edition)’s Table 13.4 as a single use, I note that Gould’s Appendix indicates 4 uses, albeit I accept that not all 4 would have been FDA approved drugs. However, the emphasis placed on this factor by Bayer fails, in my view, to take into account that the Skilled Team/Formulator had to deal with a new chemical entity, different from existing drugs on the market, which might require a different or less common counterion in its formulation.
iii) Although the hydrochloride salt would certainly be included, the Skilled Formulator would be aware of the risk of the common ion effect rearing its head. It would not require any invention for the Skilled Formulator to consider the possibility of the common ion effect occurring and to follow the suggestion made in Aulton also to include the tosylate and the mesylate. Again, it would take no invention for the Skilled Formulator to follow the recommendation in Gould to note the salts halfway up series 1, namely the hydrochloride, mesylate, maleate and tosylate salts, but, on the basis of his or her pKa analysis, to select the tosylate salt ahead of the maleate. Even leaving Aulton and Gould on one side, it would not take invention to select tosylate for inclusion in the salt screen, based primarily on pKa considerations. I am satisfied that, having identified tosylate as a candidate, the Skilled Formulator would not have been put off including it.
- Like Birss J in Hospira v Genentech, I am quite sure if one compared a number of real skilled teams side by side, having read Lyons and faced with sorafenib, they would select different ranges of salts to test in a first or second tier, albeit with considerable overlap. Some teams who found unpromising results in the first and second tier screen would continue past a second tier screen, others might not. I bear in mind that some real teams might never have selected the tosylate salt for inclusion (depending on their particular experience), but I am satisfied that most would. Above all, the inclusion of the tosylate salt would have been the result of standard and routine considerations.
- For all the reasons set out above, I find claim 12 of the Patent invalid for obviousness. Teva’s claim succeeds.
- First, after I had essentially completed what is set out above, Teva’s solicitors wrote to inform me that, at the conclusion of the oral hearing on 29 September 2021, the German Federal Patent Court announced its decision that claim 12 of the EP(DE) equivalent of the Patent was invalid for lack of an inventive step. That letter also reminded me that included in my trial bundles was a translation of the preliminary opinion of the Court, although, when preparing this judgment I had forgotten all about that opinion and had no regard whatsoever to it.
- As is clear from the timing, the decision of the German Federal Patent Court had no influence on my judgment, although it is pleasing to note the Courts of different EPC states reaching consistent conclusions.
- Second, in response to the circulation of my draft judgment, Bayer’s Counsel invited me to consider whether I should deal in this judgment with the obviousness allegation on the alternative construction advanced by Bayer ‘namely that the additional feature is implied into the claim’. Bayer submitted that obviousness on this alternative basis was ‘fully before the Court’ and suggested I had received full submissions on it, giving a transcript reference to Teva’s oral closing submissions.
- In response to this invitation, Teva’s Counsel disagreed that the issue was ‘fully before the Court’. I agree. I also agree with Teva that I did not receive full submissions either. The transcript reference was to a single short assertion that Teva succeeded even on the alternative basis.
- If the alternative basis had been fully in issue (instead of being raised for the first time only in oral closing submissions), I consider the cross-examination of Professor Frijlink is likely to have taken a different course: in particular, I strongly suspect that greater attention would have been paid to the measured solubility figure taken from the EMA document and whether it was comparable to the calculated solubility figures, and also to the sentence in that document quoted in paragraph 174.iv) above. Furthermore, if I had received full submissions on the alternative construction, I consider they would have covered at least the obviousness/insufficiency squeeze which was addressed briefly in Bayer’s Opening Skeleton (prepared before the scope of the case was cut down). The point, as I understand it and which Bayer had correctly discerned, was that the Patent did not identify or solve any alleged problem regarding the making or the formulation of sorafenib tosylate for oral administration. Thus, once the Skilled Formulator had, through routine steps, selected sorafenib tosylate for inclusion in his or her salt screen, the medicinal chemists would have made the substance (as I have found above). After that, the substance would have been characterised and developed into a form for oral administration, again using routine steps.
- It was because I considered obviousness of claim 12 as proposed to be amended or unamended claim 12 on the alternative construction had not been properly explored in cross-examination of Professor Frijlink or submissions that I was reluctant to express a concluded view. However, if I am obliged to form a view on the obviousness attack based on the alternative construction, I remain of the view that the claim lacked any inventive step. I distrust the comparison between the calculated solubility figure for the sorafenib free base and the measured figure for sorafenib tosylate in the EMA document, which was Bayer’s principal point in its Closing Submissions as to why the Skilled Formulator would not proceed to formulate sorafenib tosylate for oral administration after having included it in his or her salt screen and characterised that substance. That Professor Frijlink enthusiastically made that comparison was not entirely his fault because Bayer’s solicitors provided him with the measured figure for sorafenib tosylate but without providing him with a measured figure for the free base (which Bayer must surely have). That comparison seems to me to be contradicted by the sentence I have quoted from the EMA document.
- Thus, although it is clear to me that the issue on the alternative construction was not properly addressed in the evidence or in submissions, such evidence as I have suggests the Skilled Formulator would have found the tosylate salt to have improved properties over the free base, and s/he would have been able, using routine steps, to develop the sorafenib tosylate substance into a form for (and suitable for) oral administration. This finding does not introduce hindsight into the analysis. It simply relies on a scientific fact (cf Birss J. in Smith & Nephew Plc v Convatec Inc. [2012] EWHC 1602 (Pat) at [16]), which the Skilled Formulator would have found in his or her routine work when seeking to develop a suitable form of sorafenib which could be administered orally. Accordingly, I conclude that even on the alternative construction of claim 12, that claim is invalid.